Nguyen DacTrung1,2
Abstract
Nanomaterials that are multi-purpose and cost-effective will be highly desirable in next-generation nanotechnology applications. Over the past two decades, nanoscale self-assembly has been considered a promising means for engineering future nanomaterials, where the underlying structures are formed by the self-organization of building blocks, such as nanoparticles, colloids and block copolymers. Such bottom-up fabrication approaches have attracted interest from multiple disciplines including materials science, chemistry, physics, applied mathematics and computer science due to their practical importance and fundamental challenges. Central to the self-assembly techniques is to design suitable assembling units, their interaction rules and assembly pathways. In this report, examples will be given to demonstrate that computer simulation has been a powerful tool for providing not only profound insights into the complex interplay between the building blocks’ geometry and their interactions, but also valuable predictions to inspire ongoing and future experiment. Theoretical background of self-assembly processes and simulation methods and tools commonly used in self-assembly studies will be briefly discussed.
Tóm tắt
Ứng dụng công nghệ nanô trong tương lai sẽ rất cần đến các loại vật liệu nanô đa dụng và chi phí thấp. Trong hai thập kỷ vừa qua, các phương pháp sử dụng quá trình tự lắp ghép ở cấp độ nanomet (1 nm = 10-9 m) được xem như một hướng chế tạo vật liệu nanô tiềm năng, trong đó các phần tử cơ bản như hạt nanô, keo và polyme tự sắp xếp tạo thành các kết cấu nhất định. Các phương pháp chế tạo vật liệu từ dưới lên như vậy đã và đang thu hút sự quan tâm nghiên cứu đa ngành bao gồm khoa học vật liệu, hoá học, vật lý, toán ứng dụng và khoa học máy tính không những bởi tầm quan trọng thực tiễn mà còn bởi những thách thức mang tính nguyên lý của chúng. Vấn đề trọng tâm của các phương pháp này chính là làm thế nào để thiết kế các phần tử lắp ghép thích hợp, cách thức chúng tương tác với nhauvà quy trình lắp ghép. Trong báo cáo này, chúng tôi sẽ đưa ra một số ví dụ cho thấy mô phỏng trên máy tính là một công cụ mạnh mẽ mang đến những hiểu biết sâu sắc về sự ảnh hưởng qua lạigiữa hình dạng và tương tác của các phần tử lắp ghép, cũng nhưnhững dự đoán giá trí mang tính gợi mở cho nghiên cứu thực nghiệm. Chúng tôi cũng sẽ giới thiệu cơ sở lý thuyết của quá trình tự lắp ghép, các phương pháp mô phỏng và công cụ đang được sử dụng phổ biến trong lĩnh vực nghiên cứu này.
1. Introduction
As future technology is increasingly driven toward smaller length scales, shorter time scales and environmental-friendly and energy efficient operations, the need for nanomaterials that are cost-effective and reconfigurable becomes inevitably urgent. These types of nanomaterials would substantially benefit a broad range of nanotechnology applications including, but not limited to, catalysis, electronic, energy conversion and storage, optical and medical devices, and drug delivery. Material scientists have been looking for efficient, robust and versatile approaches to better control the underlying structure of nanomaterials and their mass production. Among promising approaches, self-assembly stands out as a powerful means to meet such requirements. A generic mechanism ubiquitous in biological systems, self-assembly is governed by the minimization of free energy, resulting in the spontaneous organization of nanoscopic building blocks, e.g., block copolymers, nanoparticles and colloids, into thermodynamically and mechanically stable structures. In other words, nanomaterials are assembled from bottom up, without the need of human manipulation at the nanometer scale. The fundamental challenges to self-assembly approaches, nonetheless, lie in how to design suitable building blocks,their interaction rules and assembly pathways for the target nanostructures.
The application of self-assembly to nanomaterials fabrication has been fueled by recent advances in synthetic techniques, which enable precise control over building block topology. Nanoparticles and colloids can be synthesized in various shapes, sizes, materials and compositions. They can also be functionalized with polymers and DNAs to form a vast library of “colloidal molecules” (1–11). In many cases, these colloidal molecules can even adopt multiple conformations in an analogous manner to actual molecules in response to changes such as temperature, pH and light in their surrounding environment. Additionally, the primary interactions between these nanoscopic building blocks are non-covalent in nature, such as van der Waals dispersion, electrostatics, hydrophobic/ hydrophilic forces and hydrogen bonding (12, 13). This indicates that the interaction between nano building blocks are relatively independent of their chemistry, and thus, more versatile than their atomic and molecular counterparts. As a result, theself-assembly of nanoscale building blocks has become an excellent object for simulation studies, where generic coarse-grained models are sufficient to capture the essential physics of the phenomena of interest.
Since the first studies conducted in the late 1950s, computer simulation has greatly evolved into a powerful, and in many cases, indispensable, tool for investigating atomic, molecular and mesoscopic systems. The revolutionary advances in electrical engineering and simulation algorithms over the past two decades have enabled computational scientists to look into problems on many orders of magnitude greater in time and length scales and from various angles. More interesting, perhaps, is that computational studies on self-assembly for engineering nanomaterials have become multidisciplinary, involving collective knowledge from chemistry, physics, applied mathematics and computer science. The objectives of this report are to introduce a broad picture of computational self-assembly studies to readers unfamiliar to the field, and to motivate young scientists to pursue research on this rapidly expanding, high-impact area of nanoscience. This report is necessarily incomplete, and while we make every attempt to include pioneering contributions, highly cited literature and findings from leading research groups in the field, we hope the readers forgive us for missing important references that should have been cited.
Our report is organized as follows. In Section II, we will give a brief introduction to the theoretical background, simulation methods and tools commonly used in self-assembly studies. We will show by examples in Section III how computer simulation has helped provide insights into intriguing self-assembly processes observed in experiments. Finally, computer simulation is shown to serve as an efficient design tool, providing guidance to experimental investigation (Section IV).
2. Background, methods and tools
2.1. Theoretical background
Self-assembly at equilibrium is governed by the second law of thermodynamics, whereby the system of nano building blocks evolves to minimize free energy. For instance, for a given number of building blocks at constant temperature and volume (i.e., in the canonical ensemble), the Helmholtz free energy F = E − TS is minimized, where E is internal energy, T is temperature and S is entropy (14, 15). Starting from any initial configuration, the building blocks will self-organize so that E is decreased and S is increased simultaneously. In principle, the building blocks will eventually form a thermodynamically stable structure at the given density and temperature. As can be easily seen, the competition between the energetic term, E, and entropic term, −TS, determines the stable structure. At high temperature, the entropic term dominates and the stable states are mostly isotropic, or disordered. As temperature is lowered, the energetic term becomes more influential and the building blocks tend to exhibit certain ordering at the expense of the system entropy. For self-assembly applications in soft matter systems, of which the characteristic energy scale is comparable to thermal fluctuations, the temperature range of particular interest is slightly below the disorder-order transition point.
Similar arguments on free energy minimization apply to equilibrium self-assembly under other thermodynamic constraints such as constant pressure-constant temperature (i.e., the isobaric-isothermal ensemble), and constant chemical potential-constant temperature (i.e., the grand-canonical ensemble). When assembly proceeds out of equilibrium, for instance, as induced by evaporation, flows, gravity and electric or magnetic fields, special treatments will be required (15). In many cases, where the energy scale of the external fields is comparable to that of the building block interactions, it is usually accepted that the equilibrium framework is still relevant.
There are several approaches to designing nanostructures from bottom up given a collection of assembling building blocks. In the inverse statistical mechanics approach, for instance, one attempts to find the optimal interaction between the building blocks that makes the target structure more energetically and mechanically stable than other possible competitors (16–18). The optimized interaction is then used in computer simulation to examine if the target structure can actually form. Otherwise, the interaction optimization continues with the next iteration. In this scheme, there is no guarantee that the obtained interaction would always be plausible in experiment. Another approach is to identify several candidates (often crystalline phases) which the building blocks may assemble into, and perform free energy calculations to find the thermodynamically most stable structure (19–21). Alternatively, one starts with an experimentally familiar form of the building block interactions, e.g., van der Waals and Coulombic interactions, and performs simulation with the relevant parameters such as concentration, temperature, pressure, interaction strength and building block composition varied systematically. Although the target structure may or may not be found in this forward engineering approach, arguably there are reasons that it becomes common in practice. First, the model interactions are often justified by experiment. Second, relevant physical parameters can be systematically investigated. Finally, it can provide predictions to the regions of the parameter space that have not been explored in experiment. In the following sections, simulation methods and tools routinely used for forward engineering studies will be discussed.
2.2. Simulation methods
Roughly speaking, common simulation methods for atomic and molecular systems can be classified into two categories: those that are based on Molecular Dynamics (MD) and the others on Monte Carlo (MC) methods. MD and MD-based methods such as Brownian Dynamics and dissipative particle dynamics (DPD) sample equilibrium states by integrating the Newtonian equation of motion of all the evolving atoms or particles. Whereas, MC-based methods generate trial configurations, which are then accepted or rejected based on a given probability distribution. Intermediate methods between MD and MC have also been proposed to maximize their advantages (22).
The theoretical foundation of molecular simulation methods is established by statistical mechanics (23–25), which connects macroscopic observables such as energy, temperature and pressure to statistical measurements from microscopic configurations, either by time averaging in MD simulations, or by ensemble averaging in MC simulations. The optimal choice for the simulation method is strongly dependent upon the length and time scales of the systems of interest and the objectives of specific studies. Interested readers are referred to classical textbooks such as Refs. (23–25) for thorough discussion. Beyond sampling macroscopic quantities in equilibrium states, state-of-the-art molecular simulations have been employed in studies of non-equilibrium processes and rare-event kinetics (26, 27), and in free energy calculations (28).
It is important to stress that while there is little doubt about the power of molecular simulation, practitioners should also be aware of its “dark side” (29). Finite size effects, poor statistical sampling and using flawed methods are among common factors that lead to erroneous interpretation of simulation results. Evidently, the relevance of simulation results is heavily dependent upon the model in use; whereas, the chosen simulation method determines the simulation results’ statistical meaningfulness and computational efficiency, both of which are strongly problem-specific. For example, if the goal is to predict the equilibrium structures assembled at a low temperature (or equivalently, strong attraction between the building blocks), running a traditional MD (or MC) simulation at that low temperature, however long, would give poor statistical results because the system is very likely kinetically arrested in local energy minima. Instead, one should (1) run multiple simulations from different initial configurations and (2) vary the heating/cooling schedule toward the target temperature. The former is to ensure the outcome independent of initial conditions, and the latter is to help the system escape from potential kinetic traps. Even better is to employ advanced methods such as parallel tempering (24, 28), which use MD or MC simulations as their workhorse, to accelerate the sampling process.
2.3. Simulation tools
Since the first Molecular Dynamics simulation study by Alder and Wainwright in 1957 (30), which was for 32 hard spheres in a cubic box, tremendous advances have been made to algorithms, software and computer hardware, aiming at both fundamental and practical problems. Simulations at length scales of hundreds of nanometers and time scales of nanoseconds have become routine on either commodity clusters or supercomputers. With regards to self-assembly studies, advances in simulation modeling, methods and techniques have been extensively employed for predicting equilibrium structures, characterizing their thermodynamic and mechanical stability, as well as analyzing their responses to perturbations and external fields.
The interactions between nanoparticles, colloids and block copolymers at nano- and mesoscales are generally short ranged in nature. For instance, van der Waals forces decay with r−6, where r is the distance between particles. Electrostatic interactions, often screened by medium dielectricity and counterions, decay substantially faster than r−3. The building blocks effectively interact with several adjacentneighbors. Coarse-grained models at nanoscales are therefore well suited for parallel computing, where computation is performed concurrently on independent compute units. Algorithms for parallelizing MD simulations have been proposed since the late 1980s and greatly improved since then; those for MC simulations have been few (31). Fortunately, these algorithms have been implemented in popular open-source molecular simulation codes such as GROMACS (32), AMBER (33), DLPOLY (34), MCCCS Towhee (35), LAMMPS (36), NAMD (37),Desmond (38), and recently, HOOMD-Blue (39, 40) and OpenMM(41). These packages are often results of collaborative efforts of mathematicians, physicists, chemists and computer scientists to ensure accuracy and to maximize computational efficiency simultaneously. With these rigorously tested, available-at-no-cost, regularly maintained tools, scientists are now able to conduct sophisticated research at maximized efficiency without having to reinvent the wheel.
The performance of molecular simulation codes has been remarkably boosted by the rapid development in parallel computing in the last decade. As traditional central processing units (CPUs) hit the performance wall, computers are designed to embrace accelerators such as NVIDIA’s graphics processing units (GPUs) and Intel’s Many-Integrated Core (MIC) architectures. Originally designed for computer games, GPUs are now having a considerable share in scientific computing, thanks to the availability of publicly accessible programming interfaces such as NVIDIA’s CUDA and the opensource framework OpenCL. Interestingly, the most time consuming tasks in molecular simulation, such as force and energy computation, can be re-designed to exploit the fine-grained parallelism delivered by these many-core accelerators. As a result, simulations can now be performed at rates orders of magnitude faster and on smaller-scale, more affordable workstations than they were in the 1990s. For instance, using HOOMD-Blue version 0.11.2, a simulation of a polymer melt consisting of 64000 particles (a typical size nowadays) run on a desktop computer with one Intel E5-2670 CPU and one NVIDIA Tesla K20X GPU connected via a PCIe 2.0 16x interface is an order of magnitude faster than without the GPU (39, 40).
The increased simulation rate enables scientists to investigate much larger system sizes, i.e., larger numbers of building blocks, to run much longer simulations and to launch more simulations. These mean to avoid finite size effects, and to reduce statistical errors in the time-, or ensemble-averaged, results. Consequently, the amount of data produced is proportionally increased. This puts pressure not only on data storage facilities, but also on data analysis software. For offline analysis, data is written to, and read from, hard disks, to be processed by third-party software. Popular visualization and offline analysis software, such as VMD (45) and VisIt(46), are now capable of handling big data sets in parallel across multiple compute nodes and with GPU acceleration. Meanwhile, online analysis, i.e., processing data during the course of simulation, is often supported by simulation packages, so that the amount of data written to hard disks can be substantially reduced. Also, it is not uncommon that one has to implement their own analysis for their specific needs as an extension to the package’s existing source code. By doing so, they can take full advantage of the parallelization of the host code
|
|
|
Figure 1. (A) Superstructure assembled by octopods. Top-left: an octopod-shaped particle; top-right: a linear chain assembled by interlocking octopod-shape particles. Bottom: superstructure assembled by linear chains packing side by side. Inset is the energy minimizing structure predicted by simulation. Reprint from Ref. (42) with permission. (B) Lattice assembled by rhombic nanoplates: experiment (top) and simulation (bottom). Reprint from Ref. (43) with permission. (C) Superlattice formed by polyhedral-shaped silver nanoparticles in experiment (top) and simulation (bottom). The particles are in the same colors to show the agreement between experiment and simulation. Reprint fromRef. (44) with permission. |

3. Insights into experimental findings
There have been tremendous efforts devoted to the applications of self- and directed-assembly to engineering nanostructures for mass production. Nano building blocks can be synthesized from a wide variety of materials including metals, semiconductors, polymers and block copolymers, and possible combinations (see Refs. (8)and(11) for recent reviews). Assembled nanostructures have also been reported in a broad range of complexity, ranging from classical mesophases observed with block copolymers such as cubic-centered phases, hexagonally packed cylinders, double gyroid and lamellae (see Ref. (47) for a recent review) to crystalline structures formed by faceted nanocrystals(5, 48) to superlattices formed by mixtures of colloids (49, 50) and superstructures (51–56), to name a few. Additionally, the length and time scales pertinent to the assembly process span atomic to molecular scales, i.e., from several Angstroms to tens of nanometers in length and from nanoseconds to minutes in time. The diversity in assembling systems and the large window of length and time scales are among fundamental challenges for experimental scientists to reveal the underlying assembly mechanism. Moreover, the presence of a multitude of undesirable factors such as impurities, noises, polydispersity and non-equilibrium effects may in many cases shadow the key driving forces that govern the phenomena of interest.
A common practice to identify the key factors that govern a self-assembly process involves developing coarse-grained models that capture the essential physics at the relevant length and time scales. Using these models, scientists can draw conclusions on the individual and/or collective effects of various factors by performing either molecular simulation, or stability analysis. It is important to stress that without computer simulation it is nontrivial to rationalize the formation of such distinctive complex structures at the first place. For instance, Misztaet al. used simulation to explain the hierarchical assembly of octopod-shaped nanoparticles of approximately 80 nm in size into superstructures as shown in Figure 1A (42). The particles first interlock into dimers, which are more kinetically accessible than the competing configurations as to minimize the particle-particle van der Waals interaction energy. Next, approaching particles are entropically favored to interlock into the already formed dimers at two ends, where sterical hindrance is the smallest. The linear chains subsequently pack side by side to form the final structure. In this example, nanoscale modeling and simulation serve to explain how the peculiar shape of the building blocks determines the local packing and assembly pathway toward the superstructure.
Recent experiments by Murray group found that highly faceted planar lanthanide flouridenanocrystals form long-range ordered tilings at the liquid-air interface (43) (Figure 1B). To understand the formation of such structures, the authors performed both first-principle calculations and mesoscale simulations. The former provides atomic-level details on the attractive interaction at the edges of the nanocrystals; the latter shows the interplay between patchiness and shape anisotropy that results in different packing configurations observed in experiment. As another example, silver nanoscrystals in various polyhedral shapes were experimentally shown to assemble into their theoretically predicted densest packings, except those with an octohedral shape (44) (Figure 1C). The octohedral-shaped nanocrystals form a superlattice with a previously unknown packing pattern composed of two distinct motifs: lines and counter-rotating helices. Using a coarse-grained model and Monte Carlo simulations, Henzieet al. demonstrated that it is the depletion attractions between the octohedra that are responsible for the formation of such motifs. Examples of collaborative experimentandsimulation studies on self-assembly can be found easily in the literature (53, 56, 57).
|
|
|
Figure 2. (A-D) Polyhedra in different shapes are predicted to self-assemble into various complex crystals and quasicrystals. The structures are shown to be entropically stabilized by shape anisotropy. Reprint from Ref. (60) with permission. (E) Diamond-like structure formed by patchy particles. Reprint from Ref. (61) with permission. (F) Tetragonally cylinder structure and (G) (6;6;6) columnar structure assembled by di-tethered nanospheres with different planar angles, θ, between two tethers. Reprint from Ref. (62) with permission. |

4. Predictive design and discovery
While experimental studies are constrained by available synthetic techniques, simulation studies are allowed for much flexibility in proposing models and predicting outcomes. In many cases, predictions from simulation have been successfully used to guide later experimental studies. Predictive simulation studies usually involve systematic investigation of how control factors, such as temperature, density and building block geometry, influence the assembled structures. The predictions can be summarized by “phase diagrams”, which map a pair of the control factor values such as temperature-volume fraction, or interaction strength-pressure, with the resulting nanostructures, or phases.Early examples of such phase diagrams are those of block copolymers (58) and liquid crystals (59), where the assembled structures are mapped to the pressure-density or temperature-density planes.
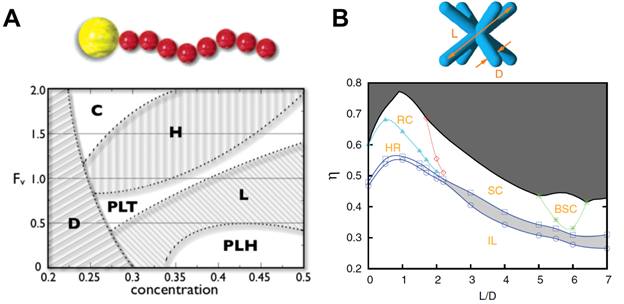
But assembled structures are not simply dependent upon thermodynamic parameters. In their seminal study, Glotzer and Solomon (63) put forward a generic scheme in which building block anisotropy would lead to novel dimensions for assembling nanostructures from bottom up. Shape, aspect ratio, patchiness and faceting are among the anisotropy dimensions along which building blocks are predicted to assemble into complex nanostructures. Examples of such assembled structures are given in Figure 2A-E. Simulation studies also proposed that by tethering nanoparticles with a finite number of immiscible polymer chains, one can assemble a wide variety of nanostructures (62, 64–69) (Figures 2F and 2G). The resulting structures are shown to exhibit notable similarity with those formed by block copolymers and those by liquid crystal molecules such as micelles, hexagonally packed cylinders, perforated lamellae and lamellar phases. It is the interplay between the incompatibility between the building block components and the local packing of the rigid groups in different shapes that give rises to such rich phase behaviors. Meanwhile, particles with sticky patches, Janus particles and faceted particles, have also been shown to form terminal clusters (61, 70) and surprisingly complex crystalline and quasi-crystalline structures (20, 60, 71, 72). Figure 3 provides two example phase diagrams obtained by computer simulation. In the first, using Brownian Dynamics simulation, Iacovellaet al. studied the self-assembly of nanospheres with one polymer into various phases including lamellae, perforated lamellae and cubic-ordered spherical micelles (66). In the second example, Qi et al. predicted numerous nanostructures formed by octopod-shape colloidal particles on a flat substrate using Monte Carlo simulation. The rhombic crystal, square-lattice crystal and binary lattice square crystal structures were already observed in experiment (57). These studies assert that the building block geometry, the number of the patches or tethers, and their relative positions and selective attraction all play sophisticated roles in determining the final structures.
To characterize the structural properties of the assembled structures, one should employ certain metrics that are representative of the ordering of the building blocks either locally or globally, or both. One of natural metrics is the system potential energy, of which a decreased value corresponding to building block aggregation. However, in most cases, potential energy tells little about how the building blocks pack within the assembled structures. More specific order parameters are needed in such cases. For example, the alignment of rod-like particles in liquid crystalline phases (e.g., nematic or smectic phases) is usually characterized by the nematic order parameter, P2, which is the largest eigenvalue of the 3 by 3 matrixpαβ= <3u(i)αu(i)β-δαβ>/2, where u(i) = (ux, uy, uz)is the unit length director of the rod-like particle i; α,β = x,y,z; δαβ is the Kronecker delta, and the bracket <·>represents the averaging over all the particles in the system. P2 may vary from zero to unity, corresponding to an isotropic phase (no alignment) and a nematic phase (perfect alignment), respectively. Combining multiple relevant metrics leads to a shape descriptor, which can be used to characterize arbitrary ordered structures (73, 74). As long as the difference between two shapedescriptor instances can be quantified, e.g., via an arithmetic operator, one can use the shape descriptor to compare the obtained structure with those in a reference library, monitor the self-assembly process toward a target structure, or automatically detect similar structures (73–75). This is a vivid example of the application of computer science knowledge (shape descriptors, data mining and machine learning) into materials science problems.
Finally, it is also interesting to note recent efforts that have focused on shape-programmable and self-propelled building blocks. Inspired by the ability of biological macromolecules to adopt various conformations in response to their environment, experimentalists look at polymer-based building blocks that deform or change shapes in a controllable manner (76–78). For example, Janus microcylinders made from poly(lactic-co-glycolic acid) (PLGA) morph into anisotropic shapes upon heating or cooling (78). Simulation studies have also predicted numerous advantages with this type of shapeshifting building blocks, including more efficient assembly and novel pathways to ordered structures (79, 80). As a result, not only does shape matter, but assembly pathways also are crucial in order to robustly obtain target structures (81). Meanwhile, self-propelled building blocks are reminiscent of active matter, where the building blocks are capable of converting input energy, e.g., in the form of light or chemical reactions, into their mobility (82). These novel building blocks allow new dimensions for self- and directed- assembly applications, and at the same time, demand novel treatments as their assembly processes take place out-of- or far-fromequilibrium.
|
|
|
Figure 3. Examples of the phase diagrams predicted by computer simulation: (A) self-assembled mono-tethered nanospheres at |

Acknowledgements
The authoracknowledges the support from the Vietnam Education Foundation, and thanks Sharon Glotzer for helpful comments.
About the author
Dr. Nguyen Dac Trung earned his BS in Energy Engineering at the Hanoi University of Science and Technology in 2004. He received his Ph.D degree in Chemical Engineering and Scientific Computing in 2011 at the University of Michigan, Ann Arbor, Michigan, USA. During 8/2011-5/2014, he worked at the National Center for Computational Sciences at the Oak Ridge National Laboratory, Tennessee, USA, as a postdoctoral research associate under the American Recovery and Reinvestment Act (ARRA) Fellowship program. His research interests include soft matter physics, interfacial phenomena, computer simulation and high-performance computing. In particular, his recent studies focus on 1) the application of self- and directed-assembly of soft matter building blocks to designing functional nanomaterials, 2) the molecular-level behaviors of confined systems, and 3) the development of GPU-accelerated force fields for molecular dynamics simulations. He was awarded the Vietnam Education Foundation (VEF) Doctoral Fellowship during 2005-2011 and the VEF Fellows and Scholars Association Scientific Award in 2012.
References
-
Breen TL, et al. (1999) Design and self-assembly of open, regular 3Dmesostructures. Science284:948–951.
-
Roh K, Martin DC, Lahann J (2005) Biphasic Janus particles with nanoscale anisotropy. Nature4:759–763.
-
Jones MR, et al. (2010) DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat. Mater 9:913–917.
-
Sacanna S, Irvine WTM, Chaikin PM, Pine DJ (2010). Lock and key colloids. Nature 464:575–578.
-
Rossi L, et al. (2011) Cubic crystals from cubic colloids. Soft Matter7:4139–4142.
-
Jiang S, et al. (2010) Janus particle synthesis and assembly. Adv. Mater 22:1060–1071.
-
Zhang Y, Lu F, van der Lelie D, Gang O (2011) Continuous phase transformation in nanocube assemblies. Phys. Rev. Lett. 107:135701.
-
Lee KJ, Yoon J, Lahann J (2011) Recent advances with anisotropic particles. Current Opinion in Colloid & Interface Science, 16:195–202.
-
Kowalczyk B, et al. (2012) Charged nanoparticles as supramolecular surfactants for controlling the growth and stability of microcrystals. Nat Mater 11:227–232.
-
Wang Y, et al. (2011) Colloids with valence and specific directional bonding. Nature 491:51–55.
-
Zhang L, Niu W, Xua G (2012) Synthesis and applications of noble metal nanocrystals with high-energy facets. Nano Today, 7:586–505.
-
Israelachvili JN (2011)Intermolecular and surface forces, Third Edition, Academic Press.
-
Bishop KJM, Wilmer CE, Soh S, Grzybowski BA (2009)Nanoscale forces and their uses in self-assembly. Small 5:1600–1630.
-
Chandler D (1987)Introduction to Modern Statistical Mechanics. Oxford University Press.
-
McQuarrie DA (2000)Statistical Mechanics. University Science Books 2000.
-
Rechtsman M, Stillinger FH, Torquato S (2005)Optimized interactions for targeted self-assembly: Application to honeycomb lattice. Phys. Rev. Lett. 95:228301.
-
Torquato S (2009) Inverse optimization techniques for targeted selfassembly. Phys Rev Lett 5:1157–1173.
-
Cohn H, KumaA (2009) Algorithmic design of self-assembling structures. Proc. Natl. Acad. Sci. USA 106:9570–9575.
-
Filion L, et al. (2009) Efficient method for predicting crystal structures at finite temperature: Variable box shape simulations. Phys. Rev. Lett. 103:188302.
-
Qi W, et al. (2013) Phase diagram of octapod-shaped nanocrystals in a quasi-two-dimensional planar geometry. J. Chem. Phys. 138:154504.
-
Dijkstra M (2013) Phase diagrams of shape-anisotropic colloidal particles. Proceedings of the International School of Physics Enrico Fermi Course CLXXXIV Physics of Complex Colloids.
-
Allen MP,Quigley D (2013)Some comments on Monte Carlo and molecular dynamics methods. Mol. Phys. 111:3442–3447.
-
Allen MP, Tildesley DJ (1989)Computer simulations of liquids. Oxford University Press, USA.
-
Frenkel D, Smit B (2002)Understanding Molecular Simulation From Algorithms to Applications. Academic Press.
-
Tuckerman MA (2010)Statistical Mechanics: Theory and Molecular Simulation. Oxford University Press.
-
Escobedo FA, Borrero EE, Araque JC (2009) Transition path sampling and forward flux sampling: Applications to biological systems. J. Phys.: Condens Matter 21:333101.
-
Allen RJ, Valeriani C, ten Wolde PR (2009) Forward flux sampling for rare event simulations. J. Phys.: CondensMatter, 21:463102.
-
ChipotC, Pohorille A (2007)Free Energy Calculations: Theory and Applications in Chemistry and Biology. Springer.
-
Frenkel D (2013) Simulations: The dark side. EurPhys J Plus 128:1–10.
-
Alder BJ, Wainwright TE (1957) Phase transition for a hard sphere system. J. Chem. Phys. 27:1208–1209.
-
Anderson JA, Jankowski E, Grubb TL, Engel M, Glotzer SC (2013) Massively parallel Monte Carlo for many-particle simulations on GPUs. J. Comput. Phys. 254:27–38.
-
Berendsen HJC,van der Spoel D, vanDrunen R (1995)Gromacs: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 91:43–56.
-
Case DA, et al. (2005) The AMBERbiomolecular simulation programs. J. Comput. Chem 26:1668–1688, 2005.
-
Smith W, Todorov IT (2006) A short description of DL POLY.Mol. Phys. 32:935–943.
-
http://towhee.sourceforge.net.
-
Plimpton S (1995). Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys 117:1-19.
-
Phillips JC, et al. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802.
-
Bowers KJ, et al. (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters.Proceedings of the ACM/IEEE Conference on Supercomputing.
-
Anderson JA, Lorenz CD, Travesset A (2008) General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 227:5342-5359.
-
HOOMD-blue. http://codeblue.umich.edu/hoomd-blue/.
-
Eastman P, et al. (2013) OpenMM 4: A reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 9:461– 469.
-
Miszta K, et al. (2011) Hierarchical self-assembly of suspended branched colloidal nanocrystals into superlattice structures. Nat. Mater 10:872–876.
-
Ye X, et al. (2013) Competition of shape and interaction patchiness for selfassemblingnanoplates. Nat. Chem. 5:466–473.
-
Henzie J, Grünwald M, Widmer-Cooper A, Geissler PL, Yang P (2012) Self-assembly of uniform polyhedral silver nanocrystals into densest packings and exotic superlattices. Nat. Mater 11:131–137.
-
Humphrey W, Dalke A, Schulten K (1996)VMD: Visual molecular dynamics. J. Mol. Graphics 14:33–38.
-
VisIt. https://wci.llnl.gov/codes/visit/.
-
Mai Y, Eisenberg A (2012) Self-assembly of block copolymers. Chem. Soc. Rev. 41:5969–5985.
-
Tang Z, Wang Y, Shanbhag S, Giersig M, Kotov NA (2006) Spontaneous transformation of CdTe nanoparticles into angled Tenanocrystals: From particles and rods to checkmarks, X-marks, and other unusual shapes. J. Am. Chem. Soc. 128:6730–6736.
-
Kalsin AM, et al. (2006) Electrostatic selfassembly of binary nanoparticle crystals with a diamond-like lattice. Science 312:420–424.
-
Dong A, Chen J, Vora PM, Kikkawa JM, Murray CB (2010). Binary nanocrystalsuperlattice membranes self-assembled at the liquid-air interface. Nat. Mater 466:474–477.
-
Alivisatos, et al. (1996) Organization of nanocrystal molecules using DNA. Nature 382:609-611.
-
Klajn R, Bishop JM,Grzybowski BA (2007) Light-controlled self-assembly of reversible and irreversible nanoparticle suprastructures. Proc. Natl. Acad. Sci. USA 104:10305–10309.
-
Hong DJ, et al. (2009) Solid-state scrolls from hierarchical self-assembly of T-shapedrod-coil molecules. AngewChemieInt Ed 48:1664-1668.
-
Srivastava S, et al. (2010) Light-controlled self-assembly of semiconductor nanoparticles into twisted ribbons. Science 327:1355–1359.
-
Maye MM, Kumara MT, Nykypanchuk D, Sherman WB, Gang O (2010) Switching binary states of nanoparticle superlattices and dimer clusters by DNA strands. Nat. Nanotechnol. 5:116–120.
-
Xia Y, et al. (2011) Self-assembly of self-limiting monodispersesupraparticles from polydisperse nanoparticles. Nat Nanotechnol. 108:580–587.
-
Qi W, et al. (2012)Ordered two-dimensional superstructures of colloidal octapod-shaped nanocrystals on flat substrates. Nano Lett. 12:5299–5303.
-
Matsen MW, Bates FS (1996) Origin of complex self-assembly in block copolymers. Macromolecules 29:7641–7644.
-
de Miguel E, Rull LF, Chalam MK, Gubbins KE (1991) Liquid crystal phase diagram of the Gay-Berne fluid. Mol. Phys.74:405–424.
-
Damasceno PF, Engel M, Glotzer SC (2012) Predictive self-assembly of polyhedra into complex structures. Science 337:453–457.
-
Zhang ZL,Keys AS, Chen T, Glotzer SC (2005)Selfassembly of patchy particles into diamond structures through molecular mimicry. Langmuir21:11547–11551.
-
Iacovella CR,Glotzer SC (2009) Complex crystal structures formed by the self-assembly of ditetherednanospheres. Nano Lett. 9:1206–1211.
-
Glotzer SC, MSolomon MJ (2007). Anisotropy of building blocks and their assembly into complex structures. Nat. Mater 6:557–562.
-
Zhang ZL, Glotzer SC (2003) Tethered nano building blocks: Toward a conceptual framework for nanoparticle self-assembly. Nano Let.t 3:1341–1346.
-
Horsch MA, Zhang ZL, Glotzer SC (2005) Self-assembly of polymer-tethered nanorods. Phys. Rev. Lett. 95:056105.
-
Iacovella CR, Horsch MA, Zhang ZL, Glotzer SC (2005) Phase diagrams of self-assembled mono-tethered nanospheres from molecular simulation and comparison to surfactants. Langmuir 21:9488-9494.
-
Iacovella CR, Keys AS, Glotzer SC (2011) Self-assembly of soft-matter quasicrystals and their approximants. Proc. Natl. Acad. Sci. 108:20935–20940.
-
Nguyen TD, Zhang ZL, Glotzer SC (2008) Molecular simulation study of self-assembly of tethered V-shaped nanoparticles. J. Chem. Phys. 129:244903.
-
Marson RL, Phillips CL, Anderson JA, Glotzer SC (2014) Phase behavior and complex crystal structures of self-assembled tethered nanoparticle telechelics. Nano Lett. 14:2071–2078.
-
Zhang ZL, Glotzer SC (2004) Self-assembly of patchy particles. Nano Lett. 4:1407–1413.
-
Engel M, Damasceno PF,Glotzer SC (2012) Crystalline assemblies and densest packings of a family of truncated tetrahedra and the role of directional entropic forces. ACS Nano 6:609– 614.
-
Vissers T, Preisler Z, Smallenburg F, Dijkstra M, Sciortino F (2013) Predicting crystals of Janus colloids. J. Chem. Phys. 138:164505.
-
Keys AS, Iacovella CR, Glotzer SC (2011). Characterizing structure through shape matching and applications to selfassembly. Annu. Rev. Condens. Matter Phys. 2:263– 285.
-
Keys AS, Iacovella CR, Glotzer SC (2011)Characterizing complex particle morphologies through shape matching: Descriptors, applications, and algorithms. J. Comput..Phys. 230:6438–6463.
-
Phillips CL, Voth G (2013) Discovering crystals using shape matching and machine learning. Soft Matter 9:8552–8568.
-
Sidorenko A, Krupenkin, Taylor S, Fratzl P, Aizenberg J (2007) Reversible switching of hydrogel-actuated nanostructures into complex micropatterns. Science 315:487–490.
-
Ohm C, et al. (2011) Microfluidic synthesis of highly shapeanisotropic particles from liquid crystalline elastomers with defined director field configuration. J. Am. Chem.Soc. 133:5305–5311.
-
Lee K, et al. (2012) Spontaneous shape reconfigurations in multi-compartmentalmicrocylinders. Proc. Natl. Acad. Sci. USA 109:16057–16062.
-
Nguyen TD, Glotzer SC (2010) Reconfigurable assemblies of shape-changing nanorods. ACS Nano4:2584–2594, 2010.
-
Nguyen TD, Jankowski E, Glotzer SC (2011) Self-assembly and reconfigurability of shape-shifting particles. ACS Nano 5:8892–8903.
-
Jankowski E, Glotzer SC (2012) Screening and designing patchy particles for optimized self-assembly propensity through assembly pathway engineering. Soft Matter 8:2852–2859.
-
Marchetti MC, et al. (2012) Soft active matter. arXiv:1207.2929(cond-mat.soft).


Add new comment