Anh Tuan Tran1, Ly Le2, and Bao The Pham1
Abstract
The high mutation rate of several viruses is one of the main reasons leading to dangerous epidemics or pandemics. Therefore, there is a demand for accurate prediction of dangerous mutations which lead to new phathogenic strains that resist to current drugs and vaccines. In this research, we proposed novel methods for mutation prediction based on Naïve Bayes Classifier and conducted experiments for Neuraminidase and Hemagglutinin on influenza A virus. Our method gives considerable accuracy, also matching score. These results show that our predicted sequences have high likelihood of being the same with protein families in structures and provide useful information for antiviral drug design.
Introduction
The rapid evolution rates of several viruses, such as HIV and influenza, cause threats to the health care system in the world (1-4). These species have high error (mutation) rates during reproduction process (transcription or DNA duplication), that make the variety of different genotypes. The advantage of high error rates is to give the viral abilities to adapt to new environments by escaping our immume system and resisting to current drugs. The mutations in protein sequences lead to the changes of respective polypeptide chains and protein structures. Consequently, designing drugs or vaccines counteracting these viruses becomes a challenge for biologists. Moreover, excluding natural hosts, these viruses possibly infect and cause severe diseases in other hosts. Since 1977, for example, there have been numerous report avian or swine influenza viruses infecting humans (5-8).
A mutated genotype can be a synonymous version of the original genome that seems not to influence the respective proteins, but in case of non-synonym, the mutation significantly contributes to the evolution of protein sequences and structures. Only mutated proteins surviving under selection pressure can make species evolve and adapt to new environments.
Mutations occur randomly and unpredictably, but the mutations surviving under selection possibly have predictable pattern. As far as we know, protein sequences evolve more rapidly than structures (9). This may be the result of that protein structures assign protein functions. A mutated sequence which causes a change into structure may influences protein’s function, so an individual having a mutated protein sequence cannot live as normal. Thus, we can expect that surviving mutated proteins must reserve the protein structure and function.
There are some tools relating to this subject has been developed, especially in correlated mutation prediction and protein residues-residues contact map prediction (10-16). Scientists also interest in analysis of mutation impact on protein (17, 18). However, we probably cannot find any related researches directly predicting protein mutation. These researches do not indicate what mutants occur in future. In this paper, we investigate how to predict specific protein mutations which survive under natural selection, in future, from protein family data of sequences.
Protein sequences set which is mainly-used data for this study are non-orderable discrete data type. Therefore, a statistical learning method, like Naïve Bayes Classifier, is appropriate with this problem (19). These data sets are usually big with thousands of instances and hundreds of attributes. While other complex accurate learning methods take long execution time, Naïve Bayes Classifier can learn data during an acceptable period, with reasonable accuracy. Hence, we proposed a method for predicting protein mutation which surviving under natural selection pressure by using Naïve Bayes Classifier and also conduct experiment for Membrane Glycoproteins of Influenza A Virus in Viet Nam.
Methodology
Our method included four main steps which were preprocessing, multiple sequences alignment, target determination, and mutation prediction. The overall procedure was shown in Figure 1.
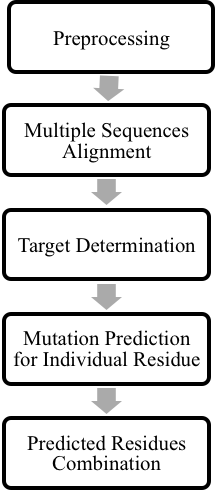
Figure 1. Overall methodology
Preprocessing
The first step we automatically eliminated sequences which contained strange characters, Algorithm 1. There are several sequences that were incomplete and had uncommon amino acids, so it was necessary to preprocess data in order to reduce noise.
Algorithm 1. Preprocessing

Multiple sequences align ment
Protein sequences in data set were not equal in length, so we had to normalize the data to the same length. Multiple sequences alignment using dynamics programming algorithm is time consuming (20). Therefore, we used progressive method with join-neighbor algorithm and Jukes-Cantor evolution distance for solving this task (21-24).
Target determination
According to Mitchel, the ith protein sequence in data set P was called an instance (sample) and denoted by pi; then, the jth position 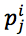 of pi is an attribute (19). In addition, we needed to determine the target ti with respect to each instance pi. We proposed three methods:1)The first method is based on phylogenetic tree, 2)The second method is based on the order of protein sequence.3) The third method is a combination of both two above methods.
of pi is an attribute (19). In addition, we needed to determine the target ti with respect to each instance pi. We proposed three methods:1)The first method is based on phylogenetic tree, 2)The second method is based on the order of protein sequence.3) The third method is a combination of both two above methods.
Phylogenetic tree
We assumed that phylogenetic trees can accurately reflect evolutionary processes of protein families. Then, we could learn rules of protein families and predict surviving sequences occurring in future, through these processes and phylogenetic trees. Target for each sequence is the sequence’s child, equation (1)
|
|
|
(1) |
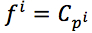
where  are children of pi.
are children of pi.
We constructed binary guide trees by using join-neighbor algorithm, Jukes-Cantor evolution distance and dynamics programming for pair wise alignment (20, 24, 25). From guide trees, we constructed phylogenetic trees by determining father for each pair of sequences which have the same higher level node, Algorithm 2.
Algorithm 2. Phylogenetic tree construction

Protein sequences’ order and component
As structural evolution rate of a surviving sequence is lower than its evolution rate of sequence to maintain its function. Thus, we assumed that the order and components of sequence contain information about structure. We could predict future sequence having the same structure with protein family’s structure. Target for each sequence were determined by equation (2).
|
|
si = pi |
(2) |
Mutation prediction
For each position in protein sequences, we generated a learner based on Naïve Bayes Classifier to predict mutation at this residue. After that, we combined all predicted mutation position to get the final mutated sequence. Given a protein sequence pi, the probability that the jthresidue mutates into a specific amino acid 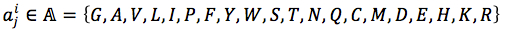 was expressed by (3), based on Bayesian formula, where
was expressed by (3), based on Bayesian formula, where  was the kth element in the possible amino acid set
was the kth element in the possible amino acid set  .
.
|
|
|
(3) |
We assumed that attributes of each instance are independent. Then, the probability could be rewritten as (4).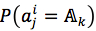 and
and 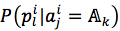 were given by (5) and (6), respectively
were given by (5) and (6), respectively
|
|
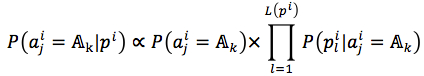 |
(4)
|
|
|
 |
(5)
|
|
|
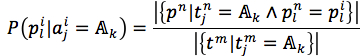 |
(6) |
where |.| is number of member in a set, L(pi) = |pi|is length of string, t is a target (t= f in case of the first method and t=s in the second method).
The mutated jthresidue was predicted by equation (7).
|
|
|
(7) |
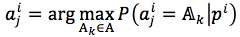
Finally, the predicted mutant of pi was  .
.
Denote that  and
and 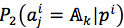 are the probability with respect to the first and the second method, according to the third method, the mutated jth residue was predicted by equation (8)
are the probability with respect to the first and the second method, according to the third method, the mutated jth residue was predicted by equation (8)
|
|
|
(8) |
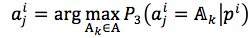
where
|
|
|
(9) |
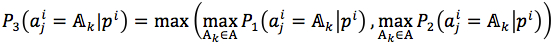
Experiments and results
Data collection and preprocessing
We collected sequences of Neuraminidase (NA) and Hemagglutinin (HA) of influenza imported from Vietnam from National Center of Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/), in FASTA format. We also collected sequences from Southeast Asia for testing purpose. After preprocessing, we obtained Table 1.
Table 1. Data selection and preprocessing
|
Region
|
Type of protein
|
Raw
|
Preprocessed
|
|
Viet Nam
|
NA
|
442
|
392
|
|
HA
|
373
|
348
|
|
|
Southeast Asia
|
NA
|
2367
|
2294
|
|
HA
|
2337
|
2248
|
1.1. Results
Firstly, we defined two evaluation criteria which are accuracy and matching score. The accuracy of our method was expressed by mean of accuracy rate of each predicted position  as equation (10)
as equation (10)
|
|
|
(10) |
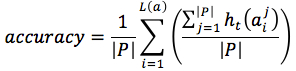
where
|
|
|
(11) |
We also computed matching score between predicted sequences and reference sequences. Gaps in predicted sequences were eliminated; then, each sequences was aligned with all reference sequences and computed Jukes-Cantor distance to each sequence in reference set. Denoted that the nearest aligned reference sequence to aligned ai was ri, the matching score was defined by equation (12)
|
|
|
(12) |
where
|
|
|
(13) |
In this research, we used NA and HA data set from Vienamfor training and testing process due to the lack of data. Table 2 describes the accuracy of our methodology. As regards NA, the accuracies of the first and the second method are almost the same, 67.87% and 65.42% respectively. Which respect to HA, the difference between two methods is just 4.58%. We also investigated the correlation between the conservation of protein family and the accuracy. As Figure 1 illustrated, almost high conservation positions ( 80%) gave high accuracy (
80%) gave high accuracy ( 80%).
80%).
Table 2. Accuracy
|
Type of protein
|
First method
|
Second method
|
|
NA
|
67.87%
|
65.42%
|
|
HA
|
73.59%
|
78.17%
|
Figure 2. Correlation between accuracy and conserved positions. (A) The first method for NA. (B) The second method for NA. (C) The first method for HA. (D) The second method for HA.
Countries in Southeast Asia are similar to Viet Nam in terms of environment, biological resources, etc., so we considered NA and HA data set in Southeast Asia as a reference set, in order to evaluate matching score. As Table 3 described, regarding NA, both three methods give moderate matching scores, 69.98%, 66.35%, and 69.17% respectively. In HA set, the accuracies of three methods are considerable, 77.92%, 78.17%, and 78.49% respectively. In addition, we randomly pick up a predicted sequence with respect to two methods and protein type to predict 3d structure (26-29). According to
Table 4, modeled positions rates are higher than 80% in both six cases.
Table 3. Matching score
|
Type of protein
|
First method
|
Second method
|
Third method
|
|
NA
|
69.98%
|
66.35%
|
69.17%
|
|
HA
|
77.92%
|
78.17%
|
78.49%
|
Table 4. Percentage of residues were modeled
|
Type of protein
|
First method
|
Second method
|
Third method
|
|
NA
|
83% (4b7qA)
|
82% (3tiaA)
|
83% (4gzoA)
|
|
HA
|
91% (2wr0A)
|
89% (2yp7A)
|
89% (2yp7A)
|
Conclusions
We proposed a novel method for predicting mutated protein surviving under selection pressure, with reasonable accuracies and matching scores, but we almost could not find out any related researches in order to compare. We did not expect a perfect accuracy because it will not suggest any novel mutated sequences. Therefore, the results are reasonable, although both two methods give under 80% of accuracies. Moreover, our method gives high accuracies with respect to conserved position which play important role in structure and function of protein. The matching score is more important than the accuracy since it indicated how our predicted sequences similar to the sequences in reference set. The more predicted sequences similar to reference set, the more their structures have high probability of resembling structure of protein family. In our research, matching scores just reach moderate levels, but the accuracy is expected to increase when using bigger reference sets. Additionally, the proportions of modeled positions are higher than 80%. These results show that our predicted sequences have high likelihood of being the same with protein families in structures.Othereffective machine learning methods are suggested to apply for the same data sets to improve accuracy and matching score. After that protein structure of predicted sequences can be constructed using homology modelling for further study.


Add new comment