Tue T. Nguyen, Tuan L.A. Pham, Tu C. Ho, Dat Q. Tran, Thinh H. Tran, Van T. Ta, and Khanh V. Tran
Center for Gene-Protein Research, Hanoi Medical University, Hanoi, Vietnam
Abstract
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism. It is caused by defects of ATP7B encoding a copper transporting P-type ATPase. The gene is located on chromosome 13q14.3. Mutation of this gene disrupts copper homeostasis, resulting in the accumulation of copper in the liver, brain, kidneys and corneas and copper toxication at these sites. Until today, more than 300 disease-specific mutations have been identified. To detect mutations of the entire ATP7B gene in Vietnamese patients with WD, sequencing analysis was applied. We detected three mutations in ATP7B genes that have not previously been reported. Two are missense mutations and one is a non-sense mutation. Furthermore, segregation analysis showed no mutation transmission patterns within each family of WD patients. Novel mutations in ATP7B in Vietnamese WD patients merit inclusion into the genetic epidemiology and the population genetic public database.
1. Introduction
Copper is an essential component of many enzymes such as: lysyl oxidase, superoxide dismutase, dopamine-β-hydroxylase and cytochrome C oxidase. These copper-dependent enzymes are needed for diverse process of oxidase metabolism including respiration, free-radical detoxification, neurotransmitter synthesis, maturation of connective tissue and iron uptake (1-2). However, copper is only required in trace amounts; accumulation of copper can damage plasma membranes, peroxisomes, mitochondria, microtubules, enzymes and even DNA (3). The effect of copper imbalance can be described best in two human genetic disorders: Wilson’s Disease (WD) and Menkes’s Disease. WD is an inherited autosomal recessive disease first described in 1912 by Samuel Alexander Kinnier Wilson. Prevalence of the disease is approximately 1 in 30,000. In 1993, Bull et al. identified the genetic defect of WD within the copper-transporting P type ATP7B gene located in chromosome 13 (4). According to Wilson’s Disease Mutation Database (http:// www.wilsondisease.med.ualberta.ca/database.asp), more than 300 disease-causing mutations have been detected and the number is still growing because mutations in the ATP7B gene vary in different populations. A recent study identified a mutation hotspot in exon 2, 8, and 10 within a sample of Northern Vietnamese population (the data have been submitted for publication). In this study we have identified three novel mutations in ATP7B with WD by using DNA sequencing. All of them are heterogeneity mutation.
2. Materials and methods
2.1. Subjects and DNA extraction
Three Vietnamese patients from unrelated families were selected. All patients had been diagnosed with WD and treated at the National Hospital of Pediatrics in Hanoi, Vietnam. WD diagnosis was based on multiple clinical symptoms, including liver failure and/or neurological issues and/or the presence of Kayser- Fleischer ring in eyes, and biochemical markers, such as low serum ceruloplasmin (CP <20 mg/dL) and high level of urinary copper (Cu >100 mg/24h) (5). Genomic DNA was extracted from peripheral blood collected in EDTA tube using Wizard Genomic DNA Purification Kit (Promega, Madison, Wisconsin, USA) following manufacturer’s protocols.
2.2. Polymerase Chain Reaction (PCR)
Entire exons of ATP7B gene were successfully amplified by using 24 primer pairs (four primer pairs for exon 2, one primer pair each for others) (IDT, Coralville, Iowa, USA). PCR was performed using GoTaq Green Master Mix (Promega, Madison, Wisconsin, USA) with 100 ng of genomic DNA in a mix containing 1U of Taq Polymerase, 10 pmol of each primer and distilled water. The thermocycle program consisted of an initial denaturation at 95 ºC for 5 min, followed by 35 cycles at 95 ºC for 30 s, 55 ºC for 45 s, and 72 ºC for 30 s, with a final extension at 72 ºC for 3 min. The size and quantity of PCR products were verified by electrophoresis in 1.5 % agarose gel.
2.3. DNA sequencing
PCR products were directly sequenced using BigDye Terminal Kit (Amersham, UK) and Avant 3100 Genetic Analyzer automated sequencer (Applied Biosystems Inc., Foster City, California, USA), and data was analyzed using CLC Genomics Workbench (CLC Bio, Qiagen, Venlo, Limburg, Germany). All products were sequenced in both forward and reverse strands.
3. Results
All three patients demonstrated similar symptoms and laboratory analyses revealed low ceruloplasmin, low serum copper and increased urinary copper excretion (Table 1). Genomic DNA was isolated from the peripheral blood leukocytes of the subjects. DNA sequencing analysis was applied to identify mutations and polymorphisms of all 21 exons of ATP7B gene in 3 Vietnamese patients with WD. One nonsense mutation and two missense mutations in Table 2 were found to be novel: c.3233C>G; c.2943T>G; c.2705T>C (Figure 1). This procedure used Polyphen, an automatic tool predicting possible impact of amino acids substitution on the structure and function of human protein in analyzing the novel missenses mutation. The mutations were correctly identified as novel mutations in the WND database (http:// www.wilsondisease.med.ualberta.ca/database.asp).
Table 1: ATP7B genotype, clinical and laboratory findings
|
Mutation |
Patient |
Affected organ |
Laboratory finding |
|||
|
Liver |
Brain |
KF-ring |
Serum CP (mg/dL) |
Urine Cu (μg/day) |
||
|
c.2705T>C |
WD 52 |
- |
+ |
|
6.0 |
325 |
|
c.2943T>G |
WD 27 |
+ |
- |
+ |
<10 |
220 |
|
c.3233C>G |
WD 23 |
+ |
- |
+ |
<10 |
180 |
|
Nucleotide change |
Exon/intron |
Codon change |
Mutation |
Type |
|
c.2705T>C |
11 |
L902P |
novel |
missense |
|
c.2943T>G |
13 |
I981S |
novel |
missense |
|
c.3233C>G |
14 |
Y1078X |
novel |
nonesense |
Clinical manifestation of three patients showed involvements of three affected organs including liver, brain, and cornea with low level of serum ceruloplasmin (Table 1). Segregation analysis within 3 families (including parents and one brother) was performed for 3 patients whose mutations in the ATP7B gene had been detected, but no mutations were identified.
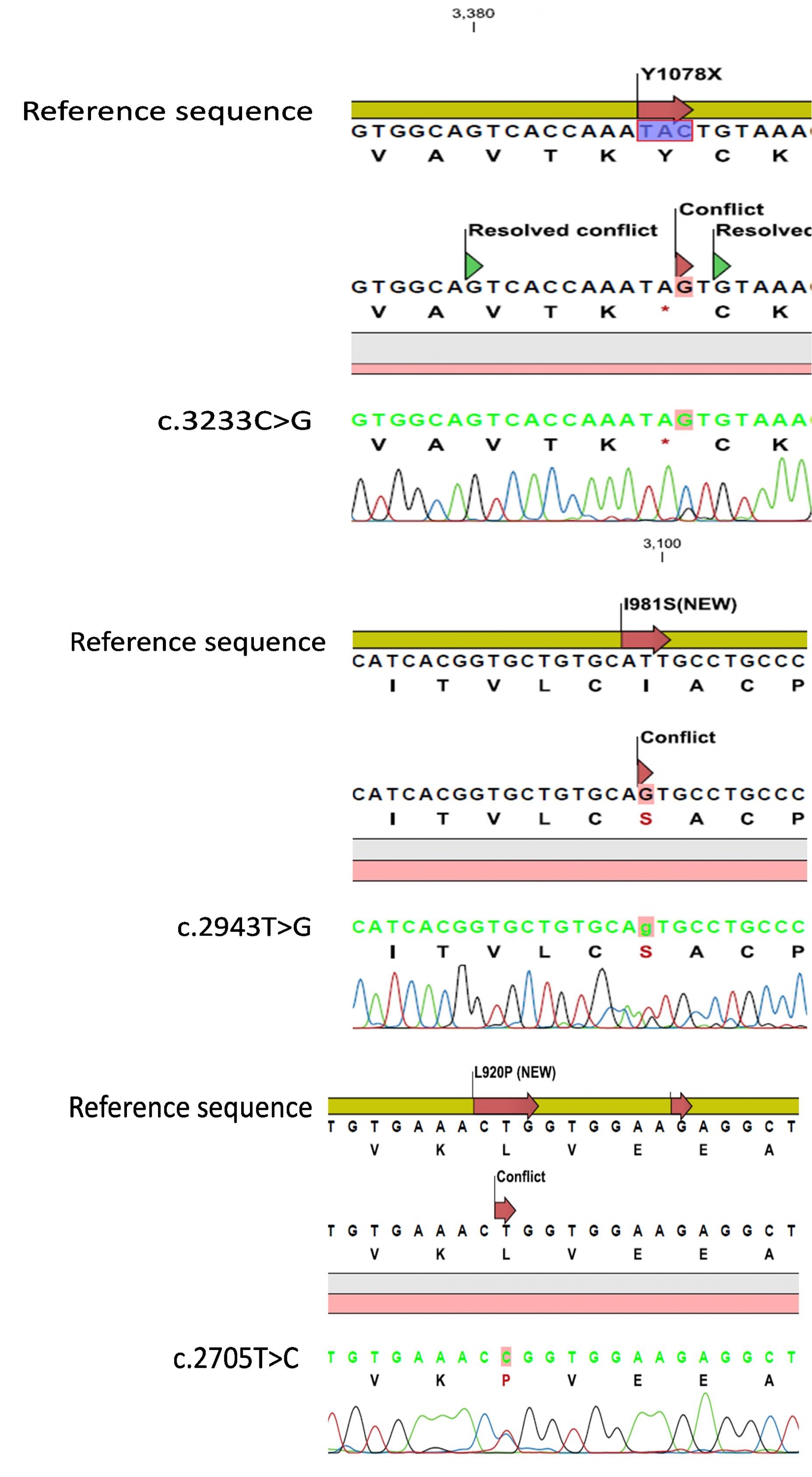
Figure 1. Sequencing results of three novel mutations on ATP7B gene of Wilson’s disease patients
4. Discussion
Wilson disease is an infrequent disorder worldwide in almost all populations. Frequencies of 1:30,000 to 1:40,000 are usually quoted (6), except for Crete and Sardinia with 6:100 and 1:7,000 live births respectively (7-8). At present, diagnosis of WD is extremely difficult especially for clinically asymptomatic individuals or those with atypical clinical manifestations. Moreover, different WD patients have different clinical features (9). Particularly, WD is representative of a low number of diseases that difficult to be treated; however, early diagnosis increases the probability of recovery (9).
Until now, more than 300 mutations have been reported in many different populations but a few of them show relative high frequencies in some geographic areas. The most frequent mutation in Europe is H1069Q with values over 65 % (10), in Sardinia, c.-441_- 427del accounts for 61.5 % of mutated chromosomes (8), mutation M645R is present in 55 % of patients in Spain (11), and R778L is very frequent in Hong Kong patients (12). Deletion c.3402delC at exon 15 has been reported worldwide at low frequency (usually below 2 %) in several populations; contrarily a high frequency (34.8 %) occurs among Brazilian index cases (13) and the most frequent mutation in Vietnam is S105X with 21.05 % (data submitted for publication).
In recent years, assisted by the development of molecular analysis, the diagnostic rate has been improved. However, clinical laboratory molecular diagnosis is expensive because there are many types of ATP7B gene mutations. Novel mutations appearing in some populations or races are inevitable; therefore, wide publication of these novel mutations and screening to identify some mutation hotspots are essential.
5. Conclusion
In our study, three novel mutations (c.3233C>G; c.2943T>G; c.2862A>G) in the ATP7B gene were detected in 3 WD patients by sequencing all exons. Our data enrich the database of Vietnamese WD as well as the WND database.
Acknowledgment
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106.16-2011.01.
References
---
Revised on 6/14/2016

