Tung Hoang1 and Gregory Dudley1*
1Khoa Hoá và Sinh hoá, Trường Đại học Florida State, Tallahassee, Florida, USA
Biên tập viên: Lâm Thiều, Virginia Polytechnic Institute and State University, Blacksburg, Virginia, USA
* Độc giả có thắc mắc về bài báo xin liên hệ email: gdudley at chem.fsu.edu
Tóm tắt: Hiểu biết về những phản ứng cơ sở trong hoá học có ý nghĩa thiết yếu để có thể áp dụng chúng cho những chuyển hoá cần thiết hay để sáng tạo ra phản ứng mới. Ở đây chúng tôi tổng kết phản ứng tách phân mảnh với các điểm nhấn về lịch sử, cơ chế phản ứng và các ứng dụng trong tổng hợp hữu cơ. Trong số các ứng dụng đa dạng này, phản ứng tách phân mảnh cho hiệu quả tốt nhất trong việc tổng hợp các hệ vòng cỡ trung bình, thường khó được điều chế qua các phương pháp khác.
Abstract: Understanding fundamental reactions in chemistry is critical essential in order to apply them for designed transformations or to invent new reactions. Here, fragmentation reactions are reviewed with emphasis on their history, mechanism and applications in organic synthesis. Among the variety of applications, fragmentation is most powerful in the synthesis of medium-sized ring systems that are difficult to prepare via alternate routes.
Từ khoá: phản ứng tách phân mảnh, tổng hợp hữu cơ.
Giới thiệu
Tổng hợp hữu cơ là một ngành xương sống của hoá học có ứng dụng rộng rãi trong nhiều khoa học liên ngành khác như khoa học vật liệu, sinh hoá, y học và đặc biệt là trong điều chế dược phẩm. Một trong các chủ đề nghiên cứu trọng điểm của tổng hợp hữu cơ là nghiên cứu tạo liên kết giữa nguyên tử các bon với một nguyên tử các bon khác (liên kết C-C) hoặc tạo liên kết giữa nguyên tử các bon với nguyên tử của nguyên tố khác thường gặp trong các sản phẩm hữu cơ như oxi, nitơ, lưu huỳnh và các halogen ... Tuy nhiên không phải lúc nào việc tạo liên kết giữa hai nguyên tử theo một yêu cầu nhất định cũng có thể được thực hiện một cách trực tiếp. Trong tuyệt đại đa số các trường hợp, khó có thể tạo ra một liên kết nếu như không phá vỡ một liên kết khác. Một trong các trường hợp phá vỡ liên kết này là phản ứng tách phân mảnh, trường hợp đặc biệt của phản ứng tách, một trong các phản ứng cơ sở của hoá học hữu cơ. Trong bài viết này chúng tôi sẽ tổng kết về phản ứng tách phân mảnh, trong đó nhấn mạnh các ví dụ quan trọng và phân tích về các chiến thuật, chiến lược được sử dụng trong các ví dụ. Để xem kĩ hơn, bạn đọc có thể tham khảo các bài viết tổng kết khác về phản ứng tách phân mảnh. 1
Lược sử và định nghĩa
Phản ứng tách phân mảnh được gặp nhiều nhất là ở trong nghiên cứu về phổ khối, trong đó ion phân tử của chất mẫu bị phá vỡ, thường là theo cơ chế gốc tự do với các phản ứng đồng ly, ít khả năng kiểm soát và do đó, ít có ứng dụng trong tổng hợp hữu cơ. Trong khi đó phản ứng tách phân mảnh theo cơ chế dị ly đã được chú ý nghiên cứu và ứng dụng. Cụ thể trong lĩnh vực tổng hợp hữu cơ, theo định nghĩa của Grob , người đã nghiên cứu phản ứng tách phân mảnh một cách hệ thống thì phản ứng tách phân mảnh là phản ứng phân cắt dị ly trong đó phân tử chất phản ứng bị phân tách thành ba phân mảnh trở lên (phương trình phản ứng 1).
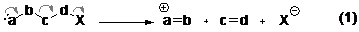
Hình 1. Phản ứng tách phân mảnh
Cùng với việc đưa ra định nghĩa cho phản ứng tách phân mảnh, Grob cũng đưa ra tên gọi cho hai trong ba phân mảnh được tạo thành sau phản ứng. Trong bài này, chúng tôi cũng sẽ dùng cách gọi của Grob cho các phân mảnh electrofuge (a=b), nucleofuge (X) và đề nghị sử dụng cách gọi phần giữa không no cho nhóm c=d. Hậu tố “fuge” có gốc từ tiếng Latinh fugere, có nghĩa là “bay đi”, có thể liên hệ với các thuật ngữ thường gặp khác là electrophile và nucleophile có hậu tố “phile” nghĩa là ‘ưa thích”. Chú ý là về mặt bản chất, nucleofuge hoàn toàn giống với nhóm rời đi trong phản ứng thế và phản ứng tách, nhưng việc gọi tên là nhóm rời đi không phù hợp trong trường hợp này, vì tuỳ theo góc nhìn mà có thể tìm thấy hai nhóm rời đi trong phản ứng tách phân mảnh: nucleofuge tách ra khỏi phần giữa không no và phần không no tách ra khỏi electrofuge.
Trong trường hợp phản ứng tách phân mảnh mở vòng, khi đó electrofuge được nối với phần giữa không no tạo thành vòng và sau phản ứng vòng này bị phá vỡ nhưng hai phần vẫn còn nối với nhau; khi đó phản ứng sẽ cho ít hơn 3 sản phẩm. Hoặc các phản ứng trong đó có thể có nhiều hơn 5 trung tâm phản ứng, như 7 trung tâm, 9 trung tâm (xem thêm chi tiết ở phần sau, mục 1.2 cơ chế phản ứng hoặc để xem ví dụ phản ứng Eschenmoser, mục 2.2.2) và sau phản ứng tạo thành nhiều hơn 3 sản phẩm, nhưng vẫn được xếp vào phân loại phản ứng tách phân mảnh.
Theo như tìm hiểu của chúng tôi trên các nghiên cứu đã được công bố, phản ứng đầu tiên có thể xếp loại phù hợp với định nghĩa của phản ứng tách phân mảnh ngày nay là phản ứng Beckman (xem mục 2.2.4): là phản ứng phân huỷ của oxim để tạo thành nitril. Một trong các ứng dụng sớm nhất và được biết đến nhiều nhất của phản ứng Beckman là trong nghiên cứu phân tích cấu trúc của morphine (Phản ứng 2-Hình 2) bằng phương pháp phân huỷ từng phần vào khoảng đầu thế kỉ 20. Khi đó, cơ chế của các phản ứng này vẫn chưa được làm rõ và do đó chưa có liên hệ gì với phản ứng tách phân mảnh.

Hình 2. Phản ứng Beckman trong phân tích cấu trúc morphine
Trong cùng khoảng thời gian đầu thế kỉ 20, một số phản ứng tách phân mảnh theo cơ chế cacbocation , trong đó sản phẩm của phản ứng thường là khó kiểm soát, chỉ được nhắc đến như là phản ứng phụ trong các quá trình khác đã được báo cáo một cách riêng lẻ, và chưa có hệ thống. Cho đến năm 1952 thì Eschenmoser lần đầu tiên giới thiệu một phản ứng trong đó chất phản ứng được thiết kế với đầy đủ các thành tố để tham gia vào phản ứng tách phân mảnh (Phản ứng 3- Hình 3). Trong công bố này của Eschenmoser, ông cũng liệt kê một danh sách tổng kết các phản ứng với cơ chế tương tự đã từng được xuất bản trước đó một cách độc lập.
Ngay sau đó, hàng loạt các nghiên cứu trong lĩnh vực đã công bố những kết quả quan trọng, phải kể đến như: Henbest lần đầu tiên khẳng định yêu cầu đối song trong sắp xếp hình học của các liên kết bị phá vỡ trong những nghiên cứu phân huỷ steroid (Phản ứng 4-Hình 4).
Grob bắt đầu với những nghiên cứu trong phản ứng tách 1,4 của dihalogen vòng , và sau đó mở rộng ra là hàng loạt những nghiên cứu toàn diện và hệ thống về cơ chế phản ứng . Vì những đóng góp này mà tên ông đã được dùng để gọi tên cho phản ứng tách phân mảnh. Những nghiên cứu quan trọng trong lĩnh vực này tiếp theo gồm có: công bố đầu tiên về trường hợp tách mở rộng vòng của Stork trong điều chế cis-cyclooctene và ngay sau đó là các nghiên cứu hệ thống của Wharton về phản ứng tách phân mảnh của 1,3 diol trong mở rộng vòng đã đưa đến việc lấy tên Wharton cho phản ứng tách phân mảnh của lớp các dẫn xuất 1,3-diol . Marshall giới thiệu việc sử dụng hợp chất của boron, dithian hoặc malonate như là electrofuge đã mở rộng khả năng lựa chọn các chất phản ứng phù hợp với từng điều kiện khác nhau. Và Corey áp dụng phản ứng tách phân mảnh vào trong tổng hợp toàn phần của caryophylene ... Trên đây chỉ là những điểm nhấn trong lịch sử phát triển của lĩnh vực nghiên cứu được người viết chọn lọc lại, chắc chắn là đã không thể đề cập hết đến nhiều nghiên cứu khác do khuôn khổ bài viết.
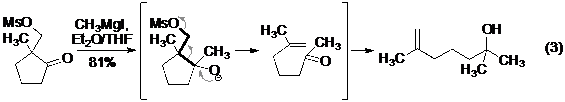
Hình 3. Phản ứng tách phân mảnh của Eschenmoser
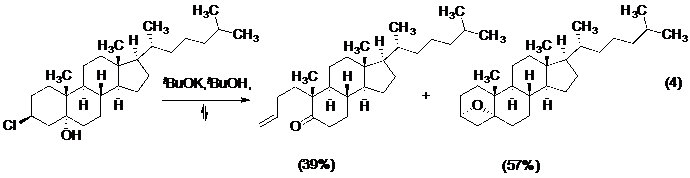
Hình 4. Phản ứng phân huỷ steroid của Henbest
Cơ chế phản ứng
Trong phần định nghĩa chúng tôi đã đề cập sơ lược đến cơ chế phản ứng để tiện cho việc phân loại một cách hệ thống phản ứng. Phản ứng tách phân mảnh có thể xảy ra theo cơ chế đồng thời hoặc từng bước. Các phản ứng tách phân mảnh đi qua trung gian mang điện âm thường đi theo cơ chế đồng thời trong khi các phản ứng tách phân mảnh đi qua trung gian mang điện dương thường đi theo cơ chế từng bước.
Phần lớn các phản ứng tách phân mảnh đi theo cơ chế đồng thời và có trung gian điện âm nên nhóm phản ứng này đã được nghiên cứu chi tiết hơn cả¬8. Ví dụ như trong phản ứng ở phương trình 1, phản ứng có 5 trung tâm và cả năm trung tâm này cùng tham gia trong trạng thái trung gian khi liên kết đôi giữa a=b, c=d được hình thành cùng lúc với liên kết đơn giữa b-c và d-X bị phá vỡ. Điều kiện tốt nhất cho phản ứng là khi chất phản ứng có thể xoay tới trạng thái lập thể có cấu hình riêng sao cho hai liên kết bị phá vỡ ở vị trí đối song. Khi đó các orbitan liên kết và orbitan phản liên kết của chúng có thể tương tác với nhau một cách hiệu quả nhất. Wharton đã dùng bốn đồng phân lập thể của decalindiol monotosylat để nghiên cứu làm sáng tỏ hiệu ứng này (Hình 5).
Cả hai đồng phân 2 và 4 của cis-decalindiol monotosylat đều có thể tham gia phản ứng tách phân mảnh để tạo thành các sản phẩm tương ứng là 5 và 6. Trong khi đó chỉ có đồng phân 1 của trans-decalindiol monotosylat là có thể phản ứng tạo ra sản phẩm 5. Đồng phân 3 có cấu trúc không thể đạt được cấu hình đối song giữa các liên kết cần phá vỡ, nên không phản ứng. Cũng tương tự như phản ứng tách, khi các liên kết bị phá vỡ ở vị trí đối song là điều kiện thuận lợi nhất cho phản ứng xảy ra. Nhưng trong trường hợp cấu trúc của chất phản ứng có cấu hình riêng cố định và không thể thoả mãn yêu cầu đối song giữa các liên kết cần phá vỡ thì phản ứng sẽ không xảy ra, như trong trường hợp trên hoặc chỉ xảy ra nếu các liên kết đó có thể đạt vị trí đồng phẳng. Holton đã chỉ ra sự quan trọng của yếu tố đồng phẳng của các liên kết cần phá vỡ trong khi nghiên cứu phản ứng tách phân mảnh của hai đồng phân endo và exo epoxi alcohol . Trong khi đồng phân endo 7a có thể tham gia phản ứng tách phân mảnh đi qua trạng thái chuyển tiếp có cấu hình syn, với các liên kết bị phá vỡ nằm ở vị trí đồng phẳng (phương trình 5-Hình 6) thì đồng phân exo 7b hầu như không phản ứng (phương trình 6-Hình 6) do các liên kết tương ứng nằm ở vị trí tương tự như vị trí gauche trong phép chiếu Newman dọc liên kết trục giữa chúng (C1-C6) và do đó các orbitan của chúng tương tác với nhau kém hiệu quả.

Hình 5. Sơ đồ nghiên cứu cơ chế phản ứng tách phân mảnh của Wharton.

Hình 6. Yếu tố đồng phẳng của các liên kết cần phá vỡ tromg phản ứng tách phân mảnh của hai đồng phân endo và exo epoxi alcohol.
Phản ứng tách phân mảnh theo cơ chế cacbocation xảy ra từng bước và có thể theo thứ tự tuỳ theo liên kết giữa nucleofuge hay electrofuge với phần giữa bị phá vỡ trước. Thường gặp nhất là trường hợp nucleofuge rời đi trước và tạo thành cacbocation trong bước đầu tiên, ion hoá. Tiếp theo sau đó là phản ứng ngược của phản ứng Prins, (Hình 7) . Do có sự tham gia của cabcocation và phản ứng Prins nên chất tham gia phản ứng theo cơ chế này thường dẫn đến sự raxemic hoá.
Ứng dụng trong tổng hợp hữu cơ
Trong quá trình tổng hợp hữu cơ, người nghiên cứu thường muốn tạo thành liên kết để xây dựng dần nên phân tử sản phẩm cuối. Vì vậy mà phản ứng tách phân mảnh, được hiểu theo nghĩa là sẽ cắt nhỏ phân tử chất phản ứng thành từng phần, thoạt nhìn có vẻ như sẽ là bất lợi cho mục đích xây dựng phân tử, thay vì phá vỡ nó. Như vậy phản ứng tách phân mảnh chủ yếu được sử dụng trong hai trường hợp: một là để xây dựng nên các nhóm chức cơ sở ban đầu, hai là để phá vỡ liên kết một cách chiến lược bởi vì tổng hợp khó có thể thực hiện qua cách khác, ví dụ như trong các trường hợp phản ứng mở vòng, hoặc phản ứng mở rộng vòng. Trong phần này chúng tôi sẽ sắp xếp các ứng dụng chọn lọc của phản ứng tách phân mảnh theo sản phẩm của phản ứng tạo thành.
Phản ứng tách phân mảnh tạo liên kết đôi
-
Phản ứng của các dẫn xuất mạch hở
Một số các nhóm bảo vệ như troc cacbonat , troc cacbamat hoặc nhóm trimethylsilyl ethyl sunfonamit , ... có cơ chế giải bảo vệ là phản ứng tách phân mảnh. Ở đây xin giới thiệu một phản ứng hữu ích trong đó Amos Smith sử dụng phản ứng tách phân mảnh của chất phản ứng không vòng kết hợp với phản ứng decacboxyl hoá của 2,3-dibromocacboxylic axit để tạo thành hợp chất 8, một chất trung gian thường gặp trong nhiều tổng hợp hữu cơ.
-
Phản ứng tách phân mảnh mở vòng
Các vòng ba hoặc vòng bốn có xu hướng mở vòng dễ dàng hơn vòng năm, vòng sáu là do sức căng của các vòng nhỏ nên cũng tham gia vào phản ứng tách phân mảnh mở vòng dễ dàng hơn. Nghiên cứu của Eschenmoser5 cũng tập trung chủ yếu vào các dẫn xuất vòng năm cạnh. Các nghiên cứu sau đó cũng chỉ ra rằng vòng sáu cạnh khó tham gia phản ứng tách phân mảnh mở vòng hơn vòng năm cạnh do có các phản ứng cạnh tranh khác. Trong một nghiên cứu gần đây Luton đã sử dụng nhóm hút electron như là ester hoặc cacbonyl ở vị trí cácbon thứ hai trên vòng bên cạnh keton để giúp làm bền hoá trạng thái chuyển tiếp của anion trong phản ứng tách phân mảnh nhằm tăng tốc độ phản ứng và đã thành công trong việc nâng cao hiệu suất của phản ứng tách phân mảnh đối với vòng sáu (phương trình 8-Hình 9). Đáng chú ý là cũng trong nghiên cứu này, tác giả đã chỉ ra rằng các dẫn xuất của iot tham gia phản ứng cho hiệu quả cao hơn dẫn xuất của brom và mesylat chứng tỏ rằng cũng có sự tham gia của nhóm nucleofuge trong giai đoạn quyết định tốc độ phản ứng.
-
Phản ứng tách phân mảnh mở rộng vòng
Do sự khó khăn của việc tổng hợp các vòng cỡ trung bình, phản ứng tách phân mảnh tỏ ra có ích đặc biệt và có lợi thế hơn hẳn so với các phương pháp khác để tạo cỡ vòng này. Đã có rất nhiều nghiên cứu đặt nền móng chi tiết cho việc tạo thành các hệ vòng cỡ nhỏ. Do đó nếu có thể sắp xếp các nhóm chức vào vị trí hợp lý để các hệ vòng này tham gia phản ứng tách phân mảnh thì sẽ rất thuận lợi trong việc tổng hợp các vòng lớn hơn, cỡ trung bình. Một số các ví dụ kinh điển của tổng hợp toàn phần rơi vào nhóm này. Trong phần trên chúng tôi đã đề cập tổng hợp toàn phần tạo nên caryophylene của Corey vào năm 1963. Gần đây Corey đã cải thiện tổng hợp này , để tạo ra sản phẩm bất đối xứng của caryophylene đi qua dienone bất đối xứng 10, lần đầu tiên giới thiệu một alken vòng bất đối xứng và ứng dụng của nó trong tổng hợp hữu cơ (Hình 10).
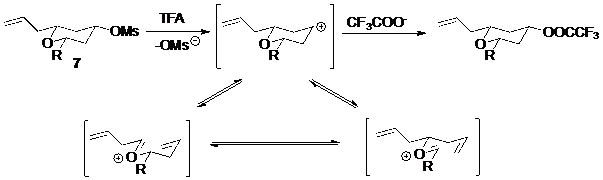
Hình 7. Phản ứng Prins, phản ứng Prins ngược và phản ứng tách phân mảnh theo cơ chế cacbocation
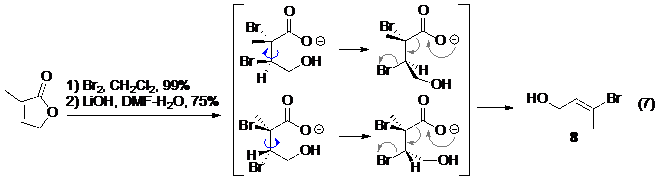
Hình 8. Một ví dụ phản ứng tách phân mảnh của chất phản ứng không vòng kết hợp với phản ứng decacboxyl hoá.
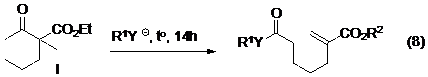
Hình 9. Nghiên cứu mở vòng sáu bằng phản ứng tách phân mảnh của Luton
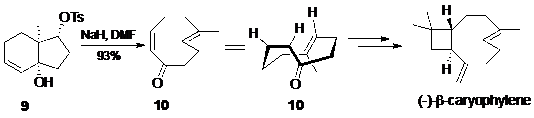
Hình 10. Sơ đồ ứng dụng phản ứng tách phân mảnh để điều chế alken vòng bất đối xứng trong tổng hợp bất đối xứng toàn phần (-)--caryophylene của Corey
Taxol và quá trình theo đuổi nghiên cứu tổng hợp toàn phần của nó có đóng góp sâu rộng trong lĩnh vực tổng hợp hữu cơ. Ở hai trong số các tổng hợp toàn phần của taxol, phản ứng tách phân mảnh đã được Holton (phương trình 9- Hình 11) và Wender (phương trình 10- Hình 11) dùng để xây dựng hệ vòng 8 ở trung tâm của phân tử, ngưng tụ cùng với một hoặc một số các vòng xung quanh. Điều thú vị là trong cả hai tổng hợp này, các tác giả đều dùng chất phản ứng là một hệ epoxi-ancol.
Một ví dụ gần đây là tổng hợp toàn phần vinigrol của Baran, trong đó nhấn mạnh sự hữu ích của phản ứng tách phân mảnh khi các phương pháp khác gặp rất nhiều khó khăn để xây dựng cấu trúc sản phẩm. Đối với vinigrol, hàng loạt các nghiên cứu và công bố của Paquette đã chỉ ra rằng việc tạo liên kết C-C để làm cầu nối hai đỉnh của hệ vòng decalin trong phân tử này là rất khó khăn và nhóm nghiên cứu của họ đã thử rất nhiều cách khác nhau nhưng đều thất bại . Chiến lược của Baran là trước hết dùng phản ứng Diel-Alder nội phân tử để tạo ra một hệ 4 vòng hợp nhất như trong 11, sau đó dùng phản ứng tách phân mảnh để cắt đứt một vòng trong số đó, tạo ra hợp chất 12 có hệ 3 vòng hợp nhất giống như cấu trúc phân tử vinigrol (Hình 12).
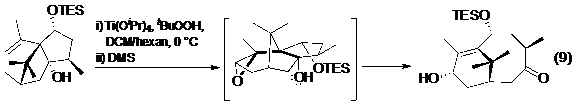
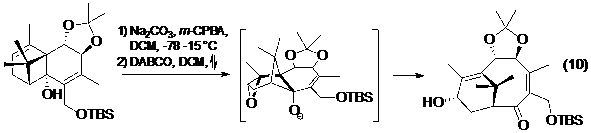
Hình 11. Phản ứng tách phân mảnh trong tổng hợp toàn phần Taxol của Holton (9) và Wender (10)
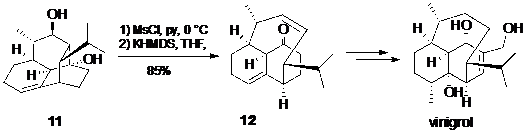
Hình 12. Sơ đồ tổng hợp toàn phần vinigrol của Baran dùng phản ứng tách phân mảnh để thiết kế cầu ansa trong hệ vòng rất khó điều chế qua cách khác.
Phản ứng tách phân mảnh tạo liên kết ba
Trong khi đã có rất nhiều các ví dụ áp dụng phản ứng tách phân mảnh để tạo thành liên kết đôi, thì lại không có nhiều như vậy các ứng dụng trong việc sử dụng phản ứng tách phân mảnh để tạo liên kết ba. Nguyên nhân có lẽ là vì yếu tố enthalpy, hơn là entropy. Vì vậy mà hầu hết các phản ứng tách phân mảnh tạo liên kết ba đều cần có các nhóm rời đi rất tốt (halogen, triflate, sunfonate), hoặc các liên kết bền hơn trong phân tử sản phẩm (như CO2, N2, N2O) để đền bù lại năng lượng tiêu hụt và làm động lực cho phản ứng. Một phương pháp thông dụng đó là kết hợp phản ứng tách phân mảnh với phản ứng decacboxyl hoá, và như vậy trong số sản phẩm phụ sẽ có CO2 với các liên kết bền của nó.
-
Phản ứng của các dẫn xuất mạch hở:
Trong các ví dụ có dùng độ bền của CO2 và N2 để thúc đẩy phản ứng, có trường hợp N2O đã được Zard sử dụng trong phương pháp điều chế ra alkyn trong mạch từ -keto este, đi qua dẫn xuất isoxazolinone. Phản ứng có một cơ chế thú vị trong đó vòng isoxazolinone trong 13 được oxi hoá bởi hỗn hợp natri nitơrit và sắt (II) sunphat trong môi trường axit sẽ tạo ra sản phẩm nitroso 14 và sau khi 14 ion hoá thành 15 thì ion lưỡng cực này tự động tham gia phản ứng decacboxyl và tách phân mảnh để tạo thành CO2, N2O và alkyn sản phẩm (Hình 13).
Trong một báo cáo gần đây, Dudley công bố phương pháp điều chế homopropargyl alcohol từ dẫn xuất vinyl triflate (phương trình 11-Hình 14). Mặc dù chất phản ứng ban đầu ở dạng vòng, nhưng sau khi đương lượng đầu tiên của nucleophil được phản ứng thì vòng này bị phá vỡ tại liên kết C-O và ở dạng mạch hở. Đương lượng tiếp theo của nucleophil tiếp tục thêm vào nhóm cacbonyl để chuẩn bị cho phản ứng tách phân mảnh, mà trong trường hợp này sản phẩm phụ là acetone, bởi vì nuclephil được dùng là methyl lithium.

Hình 13. Sơ đồ một ví dụ tách phân mảnh của dẫn xuất mạch hở bởi Zard.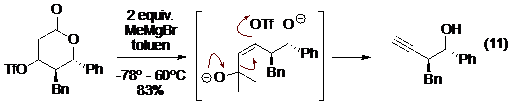
Hình 14. Điều chế homopropargyl alcohol từ dẫn xuất vinyl triflate -
Phản ứng tách phân mảnh mở vòng:
Trong nhóm phản ứng này, phản ứng Eschenmoser-Tanabe là phản ứng được nghiên cứu rất kỹ và đã có nhiều ứng dụng nhất. Năm 1967, Eschenmoser27a công bố phản ứng này ở trên tạp chí Angewante Chimie và chỉ vài tháng sau thì Tanabe27b cũng công bố nghiên cứu tương tự, một cách độc lập, trên tạp chí Tetrahedron Letters. Chất tham gia phản ứng này là epoxi sunfonylhydrazone, chất này có thể dễ dàng điều chế từ một keton không no thông qua hai phản ứng epoxi hoá và hydrazone hoá liên tiếp. Cơ chế phản ứng khá đặc biệt, trong đó có sự tham gia của 9 trung tâm, thay vì 7 trung tâm phản ứng như thường gặp và có sự sắp xếp lại các electron qua 9 trung tâm này theo hai hướng đi và về.
Do phản ứng được thúc đẩy bởi sức căng của vòng epoxi trong lần sắp xếp lại electron thứ nhất và lại có sự giải phóng phân tử nitơ rất bền vững, cùng với sự tham gia của nucleofuge là anion của một axit sunfonic hữu cơ cho nên phản ứng thường xảy ra ngay cả trong các trường hợp mà điều kiện khác gặp khó khăn. Nhờ các lợi thế này nên phản ứng đã được dùng rất nhiều trong tổng hợp hữu cơ bởi vì cả hai nhóm chức alkyn và cacbonyl đều là những nhóm chức thông dụng có thể tham gia chuyển hoá khác để đạt đến sản phẩm mong muốn. Trong một thời gian dài, phản ứng Eschenmoser-Tanabe chi phối suy nghĩ của các nhà hoá học hữu cơ mỗi khi họ muốn điều chế liên kết ba.
Dudley sử dụng triflate, là nhóm rời đi tốt nhất, để làm nucleofuge và đã áp dụng phương pháp này để điều chế alkyn gắn với các nhóm chức khác như là keton, amide, alcol, alkene và các nhóm chức khác tuỳ theo loại nucleophil được sử dụng (Hình 16). Nhóm nghiên cứu của Dudley đã ứng dụng phản ứng này trong tổng hợp toàn phần của một số hợp chất tự nhiên, trong đó cắt giảm tổng số bước tổng hợp so với các phương pháp trước đó đã sử dụng, ví dụ như trong các tổng hợp của palmerolide, illudol, hirsutene , ...
Sau công bố của Dudley sử dụng triflate làm nucleofuge ít lâu, Brewer đã sáng tạo trong việc sử dụng nitơ phân tử với mục đích tương tự. Chất phản ứng ban đầu từ một hợp chất diazo được ion hoá với SnCl4 tạo thành chất trung gian vinyldiazen để có thể tham gia phản ứng tách phân mảnh (Phản ứng 12- Hình 17).
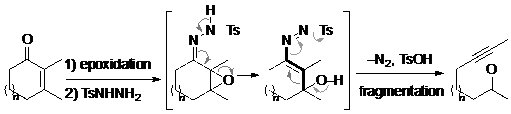
Hình 15. Phản ứng tách phân mảnh mở vòng Eschenmoser-Tanabe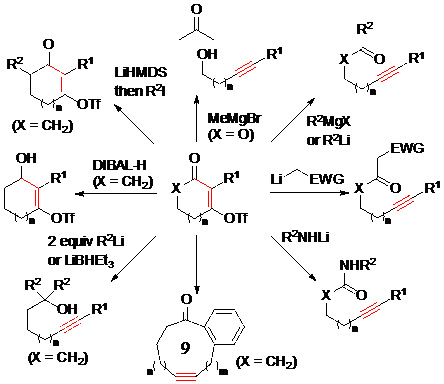
Hình 16. Ứng dụng trong tổng hợp hữu cơ của phản ứng tách phân mảnh dẫn xuất vinyl triflate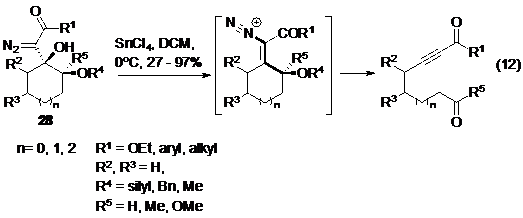
Hình 17. Ứng dụng của Brewer trong việc sử dụng nitơ phân tử làm nucleofuge. -
Phản ứng tách phân mảnh mở rộng vòng:
Tất cả các điều đã được thảo luận ở trên đối với phản ứng tạo alken vẫn hầu hết ứng dụng được cho phản ứng tạo alkyn. Tuy nhiên do các phản ứng của alkyn chưa được nghiên cứu và ứng dụng nhiều như alken, cộng với bất lợi về mặt năng lượng khi tạo ra alkyn nói chung, và nhất là alkyn ở trong vòng (do cấu tạo hình học thẳng của alkyn) nên có không quá nhiều các ứng dụng của phản ứng tách phân mảnh tạo alkyn trong tổng hợp hữu cơ như đối với alken. Và cả ba phương pháp tạo alkyne ở trên vừa nêu: phản ứng Enschenmoser-Tanabe,27 nghiên cứu của Dudley và nghiên cứu của Brewer đều có thể và đã được sử dụng trong mở rộng các vòng ngưng tụ để điều chế vòng cỡ trung bình (phương trình 13, 14, 15- Hình 18).
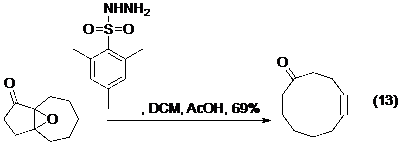
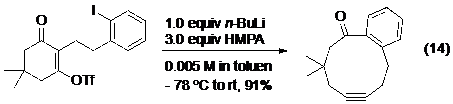
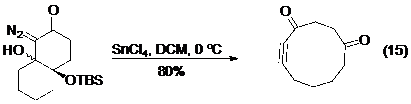
Hình 18. Ví dụ của phản ứng tách phân mảnh trong mở rộng vòng. -
Phản ứng Beckmann:
Chuyển hoá của dẫn xuất oxim để tạo ra nitril (có liên kết ba C≡N) được gọi là phản ứng Beckmann, hay phản ứng tách phân mảnh Beckmann. Grob là người đầu tiên chỉ ra rằng cơ chế của phản ứng Beckmann có liên hệ chặt chẽ với phản ứng tách phân mảnh. Và phản ứng đã được ứng dụng trong điều chế alken nối với nitril. Một trong các ứng dụng đầu tiên trong tổng hợp toàn phần là ở tổng hợp axit (±)-byssochlamic của Stork (phương trình 16-Hình 19).
Các ví dụ gần đây của phản ứng Beckmann được diễn giải ở phương trình từ 17 đến 19 (Hình 20), bạn đọc có thể xem thêm về phản ứng Beckmann trong một bài tổng kết gần đây của Hu.
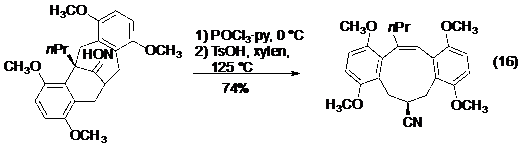
Hình 19. Ứng dụng của phản ứng tách phân mảnh Beckmann trong tổng hợp toàn phần axit (±)-byssochlamic.Bên cạnh các nghiên cứu về phản ứng tách phân mảnh tạo ra nối đôi và nối ba, các nhóm nghiên cứu của Cramer và Williams cũng đã có những công bố độc lập trong việc sử dụng phản ứng tách phân mảnh để điều chế alen.
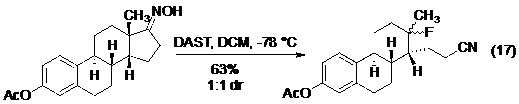
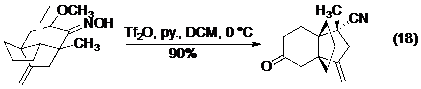
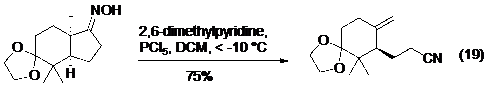
Hình 20. Các ví dụ của phản ứng Beckmann.
Tầm nhìn
Những ví dụ đầu tiên của phản ứng tách phân mảnh đã xuất hiện khá lâu, thậm chí đến hàng thế kỉ trước. Cùng với thời gian, phản ứng đã được nghiên cứu khá kĩ lưỡng về cơ chế cũng như có nhiều các ứng dụng trong tổng hợp hữu cơ. Tuy nhiên cũng có thể nhận thấy rằng cho đến gần đây vẫn còn có các nghiên cứu mới được công bố cả về phương pháp và về các ứng dụng. Có lẽ các ví dụ về ứng dụng tốt nhất của phản ứng tách phân mảnh trong tổng hợp hữu cơ nằm ở nhóm các phản ứng mở vòng và mở rộng vòng, như đã chỉ ra ở trên. Phản ứng tách phân mảnh có thể bị coi là cách gián tiếp để xây dựng cấu trúc trong tổng hợp hữu cơ, nhưng các ví dụ trên cũng cho thấy rõ ràng rằng nếu có một chiến lược phù hợp thì phương pháp này lại tỏ ra có ưu thế và hiệu quả hơn hẳn các phương pháp khác.
Tài liệu tham khảo:
* Để đảm bảo tính chính xác, chúng tôi giữ nguyên tên tiếng anh của các tài liệu tham khảo, các tạp chí và sử dụng mẫu trích dẫn của hiệp hội hoá học Mỹ.
- a) Weyerstahl, P.; Marschall, H. Fragmentation reactions. In Comprehensive Organic Synthesis, Trost, B. M., Fleming, I., Eds.; Pergamon Press: Elmsford, NY, 1991; Vol. 6, pp 1041–1070. b) Prantz, K.; Mulzer, J. Chem. Rev. 2010, 110, 3741–3766. c) Drahl, M. A.; Manpadi, M.; Williams, L. J. C–C Fragmentation: origins and recent applications. Angew. Chem. Int. Ed. 2013, in press, DOI: 10.1002/anie.201209833. d) Hoang, T.; Dudley, G. In Comprehensive Organic Synthesis, Ed 2.
- Grob, C. A.; Schiess, P. W. Angew. Chem. Int. Ed. 1967, 6, 1-15.
- Schöpf, C. Justus Liebigs Ann. Chem. 1927, 452, 211-267.
- a) English, J., Jr.; Brutcher, F. W., Jr. J. Am. Chem. Soc. 1952, 74, 4279-4282; b) Zimmerman, H. E.; English, J., Jr. J. Am. Chem. Soc. 1954, 76, 2285-2290; c) Zimmerman, H. E.; English, J., Jr. J. Am. Chem. Soc. 1954, 76, 2291-2294; d) Zimmerman, H. E.; English, J., Jr. J. Am. Chem. Soc. 1954, 76, 2294-2300
- Eschenmoser, A.; Frey, A. Helv. Chim. Acta 1952, 35, 1660-1666.
- (a) Clayton, R. B.; Henbest, H. B. Chem. and Ind. 1953, 1315-1316 (b) Clayton, R. B.; Henbest, H. B.; Smith, M. J. Chem. Soc. 1957, 1982-1993.
- Grob, C. A.; Baumann, W. Helv. Chim. Acta 1955, 38, 594-610.
- Grob, C. A. Angew. Chem. Int. Ed. 1969, 8, 535-546.
- Stork, G.; Landesman, H. K. J. Am. Chem. Soc. 1956, 78, 5129-5130.
- Wharton, P. S. J. Org. Chem. 1961, 26, 4781-4782.
- Caine, D. Org. Prep. Proc. Int. 1988, 20, 1-51.
- Marshall, J. A. Synthesis 1971, 229-235.
- a) Corey, E. J.; Mitra, R. B.; Uda, H. J. Am. Chem. Soc. 1963, 85, 362-363; b) Corey, E. J.; Mitra, R. B.; Uda, H. J. Am. Chem. Soc. 1964, 86, 485-492.
- Wharton, P. S.; Hiegel, G. A., J. Org. Chem. 1965, 30, 3254.
- Holton R. A., Kenedy R. M. Tetrahedron Lett. 1984, 25, 4455-4458.
- Jasti, R.; Rychnovsky, S. D. Org. Lett. 2006, 8, 2175-2178.
- Lee, C. B.; Chou, T.-C.; Zhang, X.-G.; Wang, Z.-G.; Kuduk, S. D.; Chappell, M. D.; Stachel, S. J.; Danishefsky, S. J. J. Org. Chem. 2000, 65, 6525-6533.
- Barlett, K. N.; Kolakowski, R. V.; Katukojvala, S.; Williams, L. J. Org. Lett. 2006, 8, 823-826.
- Cho, C.-G.; Kim, W.-S.; Smith, A. B., III Org. Lett. 2005, 7, 3569-3572.
- Hierold, J.; Hsia, T.; Lupton, D. W. Org. Biomol. Chem. 2011, 9, 783-792.
- Larionov, O. V.; Corey, E. J. J. Am. Chem. Soc. 2008, 130, 2954-2955.
- a) Holton, R. A.; Somoza, C.; Kim, H.-B.; Liang, F.; Biediger, R. J.; Boatman, P. D.; Shindo, M.; Smith, C. C.; Kim, S.; Nadizadeh, H.; Suzuki, Y.; Tao, C.; Vu, P.; Tang, S.; Zhang, P.; Murthi, K. K.; Gentile, L. N.; Liu, J. H. J. Am. Chem. Soc. 1994, 116, 1597-1598; b) Holton, R. A.; Kim, H.-B.; Somoza, C.; Liang, F.; Biediger, R. J.; Boatman, P. D.; Shindo, M.; Smith, C. C.; Kim, S.; Nadizadeh, H.; Suzuki, Y.; Tao, C.; Vu, P.; Tang, S.; Zhang, P.; Murthi, K. K.; Gentile, L. N.; Liu, J. H. J. Am. Chem. Soc. 1994, 116, 1599-1600.
- a) Wender, P. A.; Badham, N. F.; Conway, S. P.; Floreancig, P. E.; Glass, T. E.; Gränicher, C.; Houze, J. B.; Jänichen, J.; Lee, D.; Marquess, D. G.; McGrane, P. L.; Meng, W.; Mucciaro, T. P.; Mühlebach, M.; Natchus, M. G.; Paulsen, H.; Rawlins, D. B.; Satkofsky, J.; Shuker, A. J.; Sutton, J. C.; Taylor, R. E.; Tomooka, K. J. Am. Chem. Soc. 1997, 119, 2755-2756; b) Wender, P. A.; Badham, N. F.; Conway, S. P.; Floreancig, P. E.; Glass, T. E.; Houze, J. B.; Krauss, N. E.; Lee, D.; Marquess, D. G.; McGrane, P. L.; Meng, W.; Natchus, M. G.; Shuker, A. J.; Sutton, J. C.; Taylor, R. E. J. Am. Chem. Soc. 1997, 119, 2757-2758.
- Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066-17067.
- a) Paquette, L. A.; Guevel, R.; Sakamoto, S; Kim, I. H.; Crawford, J. J. Org. Chem. 2003, 68, 6096 – 6107; b) Paquette, L. A.; Efremov, I.; Liu, Z. S. J. Org. Chem. 2005, 70, 505 – 509; c) L. A. Paquette, L. A.; Efremov, I.; J. Org. Chem. 2005, 70, 510 – 513; d) Paquette, L. A.; Liu, Z. S.; Efremov, I. J. Org. Chem. 2005, 70, 514– 518.
- Boutillier, P.; Zard, S. Z. Chem. Commun. 2001, 1304-1305.
- Tummatorn, J.; Dudley, G. B. J. Am. Chem. Soc. 2008, 130, 5050–5051.
- (a) Eschenmoser, A.; Felix, D.; Ohloff, G. Helv. Chim. Acta 1967, 50, 708–713. Tanabe, M.; Crowe, D. F.; Dehn, R. L. Tetrahedron Lett. 1967, 3943–3946.
- (a) Kamijo, S.; Dudley, G. B. J. Am. Chem. Soc. 2006, 128, 6499–6507 (b) Lisboa, M. P.; Hoang, T. T.; Dudley, G. B. Org. Synth. 2011, 88, 353–363.
- (a) Lisboa, M. P.; Jones, D. M.; Dudley, G. B. Org. Lett. 2013, 15, 886–889. (b) Jones, D. M.; Lisboa, M. P.; Kamijo, S.; Dudley, G. B. J. Org. Chem. 2010, 75, 3260–3267. (c) Hoang, T.T; Dudley, G.B. Org. Lett. 2013, 15, 4026-4029. (d) Jones, D. M.; Kamijo, S.; Dudley, G. B. Synlett 2006, 936-938.
- (a) Draghici, C.; Brewer, M. J. Am. Chem. Soc. 2008, 130, 3766-3767 (b) Bayir, A.; Draghici, C.; Brewer, M. J. Org. Chem. 2010, 75, 296-302.
- Tummatorn, J.; Dudley, G. B. Org. Lett. 2011, 13, 1572–1575.
- Tsvetkov, N. P.; Bayir, A.; Schneider, S.; Brewer, M. Org. Lett. 2012, 14, 264–267
- Stork, G.; Tabak, J. M.; Blount, J. F. J. Am. Chem. Soc. 1972, 94, 4735-4737.
- Wang, C.; Jiang, X.; Shi, H.; Lu, J.; Hu, Y.; Hu, H. J. Org. Chem. 2003, 68, 4579-4581.
- Saget, T.; Cramer, N. Angew. Chem. Int. Ed. 2010, 49, 8962–8965.
- Kolakowski, R. V.; Manpadi, M.; Zhang, Y.; Emge, T. J.; Williams, L. J. J. Am. Chem. Soc. 2009, 131, 12910–12911.
Về tác giả:
Hoàng Thanh Tùng sinh ở Hạ Long một thành phố nhỏ nằm ở vùng đông bắc Việt Nam, thành phố được biết đến với bãi biển đẹp và hàng ngàn những hòn đảo nhỏ trên vịnh Hạ Long. Tùng chú ý đến hoá học từ cảm hứng của cô giáo dạy hoá cấp ba, sau đó bắt đầu theo học chương trình cử nhân hoá học tại ĐHQG Hà Nội và lấy bằng cử nhân năm 2006. Năm 2009 Tùng được nhận học bổng VEF để đi học tiến sĩ tại đại học bang Florida. Hiện tại, Tùng đang làm việc và nghiên cứu với giáo sư Gregory Dudley trong lĩnh vực tổng hợp toàn phần các hợp chất tự nhiên có hoạt
Giáo sư Gregory Dudley nhận bằng cử nhân tại đại học bang Florida năm 1995 và bằng tiến sĩ tại học viện công nghệ bang Massachusetts năm 2000 dưới sự hướng dẫn của giáo sư Rick Danheiser. Sau khi hoàn thành nghiên cứu hậu tiến sĩ được tài trợ bởi quỹ học bổng của NIH với giáo sư Samuel Danishefsky tại Học viện nghiên cứu ung thư Sloan-Kettering, ông quay trở lại đại học bang Florida và bắt đầu sự nghiệp giảng dạy ở đây dưới chức danh giáo sư thử việc từ năm 2002. Giáo sư Dudley được đề bạt chức danh phó giáo sư với biên chế dài hạn từ năm 2008 và bắt đầu nhận chức vụ phó chủ nhiệm khoa Hoá học từ năm 2012. Chương trình nghiên cứu hiện tại của ông có mục tiêu đóng góp cho quá trình điều chế dược phẩm bằng những kiến thức nền tảng của tổng hợp hữu cơ. Phản ứng tách phân mảnh có vị trí quan trọng đặc biệt trong những nghiên cứu của ông trong thời gian gần đây.


Add new comment