Thuốc kháng ung thư bằng con đường ức chế enzym Tyrosine Kinase
Đinh Châu Phi1*, Võ Đức Duy2
1 Département de Pharmacie, UFR de Santé, Université d’Angers, France
2 Department of Chemistry and Umeå Centre for Microbial Research (UCMR), Umeå University, Sweden
*Mọi thắc mắc xin liên hệ: dinhchauphi@gmail.com
Từ khóa: Tyrosine Kinase Inhibitors (TKIs), Imatinib, Ung thư bạch cầu dòng tuỷ mạn tính (Chronic Myelogenous Leukemia - CML), BCR-ABL, Ung thư dạ dày – ruột (Gastrointestinal Stromal Tumour - GIST).
Giới thiệu (Abstract)
Ngày nay, quá trình điều trị các dạng ung thư chủ yếu dựa vào các thuốc (hoá trị), các tia phóng xạ (xạ trị), và phẫu thuật. Sự kết hợp giữa việc dùng thuốc và xạ trị giúp gia tăng tác dụng của xạ trị, giảm di căn, và giữ lại được chức năng của mô chứa tế bào ung thư. Bên cạnh đó, các liệu pháp này cũng tồn tại hạn chế như phạm vi điều trị hẹp, phải điều trị nội trú, và đặc biệt là độc tính và các tác dụng phụ có hại cho sức khoẻ, ví dụ như các thuốc dẫn xuất platin gây mệt mỏi, nôn mửa, và rối loạn chức năng thận, gan, giảm hồng cầu, bạch cầu, tiểu cầu, và đôi khi có thể ảnh hưởng đến chức năng tim trong một số ít trường hợp. Chính vì thế việc phát triển các họ thuốc mới với tác dụng chọn lọc lên tế bào ung thư đồng thời có ít hoặc không có độc tính và tác dụng phụ lên tế bào lành (Targeted Therapy) đã được nghiên cứu hơn 30 năm nay. Một trong những thành quả đạt được là việc phát triển họ thuốc trị ung thư bằng con đường ức chế các enzym Tyrosine Kinase (Tyrosine Kinase Inhibitors – TKIs). Thuốc TKIs được đưa vào cơ thể bằng đường uống, do đó, bệnh nhân không cần phải nhập viện để được điều trị. Trong bài viết này, chúng tôi trình bày quá trình phát triển và hiệu quả của thuốc điều trị ung thư bằng con đường ức chế enzym Tyrosine Kinase từ thế hệ đầu tiên Imatinib (Glivec®, Novartis) được cấp phép bởi Cục Thực Phẩm và Dược Phẩm Hoa Kỳ (FDA) vào năm 2001 (với tên gọi Gleevec® cho thị trường Hoa Kỳ) để điều trị ung thư bạch cầu dòng tuỷ mạn tính. Bên cạnh đó bài viết cũng sẽ đề cập đến các thế hệ thuốc ức chế enzym Tyrosine Kinase được phát triển gần đây và có triển vọng.
Giới thiệu chung về họ enzym Tyrosine Kinase
Tyrosine Kinases (TKs) là một họ enzym được phát hiện lần đầu cách đây hơn 30 năm [1]. Tính đến nay, khoảng 58 enzym TKs đã được định danh [2] và nhiều nghiên cứu được tiến hành để tìm hiểu về hoạt động của họ enzym này. TKs đóng vai trò quan trọng trong các quá trình sinh trưởng, chuyển hóa, phân chia, và tồn tại của tế bào (Hình 1) [3]. Đột biến di truyền làm cho các enzym này gia tăng số lượng quá mức và có khả năng tự kích hoạt trong tế bào dẫn đến sự tăng trưởng và nhân đôi một cách mất kiểm soát của các tế bào liên quan [4]. Đây là tiền đề của việc hình thành các tế bào ung thư [5]. Một số nghiên cứu đã chỉ ra sự liên quan của enzym TKs với các loại ung thư như ung thư xương [6], ung thư phổi [7], và ung thư máu [8]. Trong bài viết này, chúng tôi đưa ra một số ví dụ về các loại thuốc điều trị ung thư bằng con đường ức chế enzym TKs.
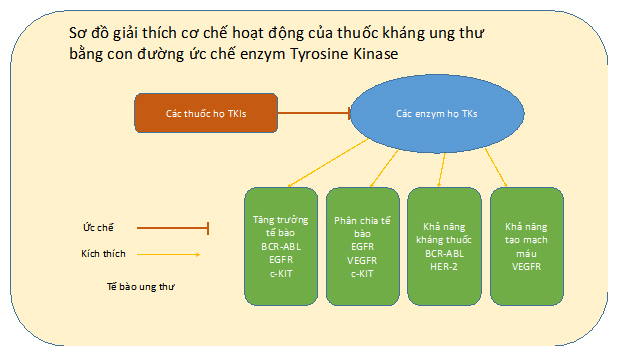
Hình 1: Khả năng ức chế enzym Tyrosine Kinase của các thuốc họ TKIs.
Thuốc Glivec® (Gleevec®)(Imatinib) trong điều trị bệnh bạch cầu dòng tủy mạn tính (Chronic Myelogenous Leukemia – CML)
Từ tuỷ xương, các tế bào tạo máu dòng tuỷ sẽ trưởng thành và phân chia thành các dạng tế bào máu khác nhau, được phát tán vào máu, hệ bạch huyết và đóng vai trò quan trọng trong hệ miễn dịch, tuần hoàn. Bệnh bạch cầu dòng tuỷ mạn tính (CML) là sự hình thành và gia tăng số lượng nhanh chóng các tế bào bạch cầu dòng tuỷ chưa trưởng thành và được phát tán vào máu dẫn đến giảm số lượng các tế bào bạch cầu trưởng thành và giảm khả năng miễn dịch. Theo thống kê của Viện Ung thư Quốc Gia Hoa Kỳ, mỗi năm, cứ 1 triệu người thì có 18 người nhiễm bệnh và 3 người tử vong vì nó. Một trong các nguyên nhân chính gây ra căn bệnh này là do sự chuyển đoạn nhiễm sắc thể t(9;22) tạo nên một nhiễm sắc thể 22 ngắn hơn bình thường. Nhiễm sắc thể này được gọi tên là nhiễm sắc thể Philadelphia (Ph) và chứa gen gây ung thư BCR-ABL trong các tế bào bạch cầu dòng tuỷ [9]. Gen này được tìm thấy trong khoảng 95% người bệnh bạch cầu dòng tuỷ mạn tính và khoảng 30% người bệnh bạch cầu cấp dòng tuỷ (Chronic Myelogenous Leukemia – AML) [10]. Gen BCR-ABL thông qua quá trình dịch mã và phiên mã (transcription and translation) sẽ tạo ra một enzym thuộc họ TKs, được gọi tên là BCR-ABL Tyrosine Kinase. Do đó, việc ức chế enzym BCR-ABL Tyrosine Kinase trở thành mục tiêu chủ yếu của việc điều trị bệnh bạch cầu dòng tuỷ mạn tính.
Từ đầu thập niên 90, các nghiên cứu đã được tiến hành để tìm ra thuốc có khả năng ức chế enzym BCR-ABL Tyrosine Kinase. Điển hình như nghiên cứu của nhóm Anafi và cộng sự dựa trên dẫn xuất của Tyrphostin [11], nhóm tác giả Mihiro căn cứ vào tác dụng của chất kháng sinh Herbimycin A [12], hay của nhóm tác giả Carlo-Stella phát triển từ hoạt chất họ flavonoid là Genistein [13].
Từ cuối thập niên 80, các nhà khoa học của hãng Novartis đã bắt tay vào nghiên cứu thuốc có khả năng ức chế enzym BCR-ABL Tyrosine Kinase. Năm 2001, thuốc Glivec® (tên khoa học Imatinib, dẫn xuất đa vòng chứa nhân pyrimidin, Hình 2) do hãng dược Novartis, Thuỵ Sĩ sản xuất đã được cấp phép bởi Cục Thực Phẩm và Dược Phẩm Hoa Kỳ (FDA) (riêng tại thị trường Hoa Kỳ, thuốc có tên gọi là Gleevec®, Hình 3) và là thuốc đầu tiên trong nhóm ức chế enzym Tyrosine Kinase được dùng cho điều trị bệnh bạch cầu dòng tuỷ mạn tính [14]. Thuốc có khả năng gây ức chế enzym BCR-ABL Tyrosine Kinase bằng cách cạnh tranh với phân tử ATP trong việc liên kết với enzym này [15]. Các thử nghiệm sinh học chứng tỏ Imatinib có tác dụng ức chế một cách chọn lọc lên các enzym ABL Tyrosine Kinases, trong đó có BCR-ABL Tyrosine Kinase. Ngoài ra, Imatinib hầu như không có tác động lên bất cứ loại enzym nào khác thuộc họ TKs [16]. Imatinib đã hoàn toàn tiêu diệt được tế bào bạch cầu ung thư trong tuỷ sau 18 tháng điều trị ở khoảng 76% trong số 1106 bệnh nhân thử nghiệm dùng, đồng thời bệnh đã không tiến triển nặng thêm trên 97% số bệnh nhân trong quá trình theo dõi thử nghiệm lâm sàng [17].
Dù các kết quả đạt được rất khả quan, Imatinib cũng tồn tại nhiều hạn chế, ví dụ tỉ lệ kháng thuốc đã tăng lên gần 50% [18]. Do đó, quá trình nghiên cứu thuốc mới ức chế enzym BCR-ABL Tyrosine Kinase đã được tiến hành và thu được những kết quả ban đầu như Dasatinib (Sprycel®, Bristol-Myers Squibb) và Nilotinib (Tasigna®, Novartis). Hai loại thuốc này có cùng cơ chế tác dụng giống như Imatinib là ức chế enzyme BCR-ABL Tyrosine Kinase và được cấp phép bởi FDA trong điều trị bệnh bạch cầu dòng tuỷ mạn tính như là thuốc thế hệ thứ hai [19–22]. Một trong hai thuốc này được dùng trong trường hợp người bệnh điều trị bằng Imatinib không mang lại hiệu quả [23–27].
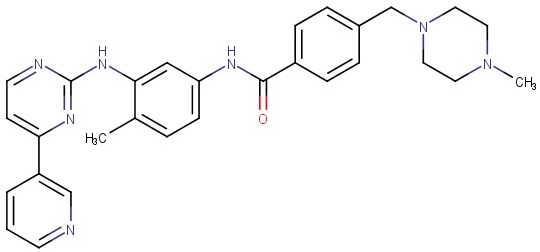
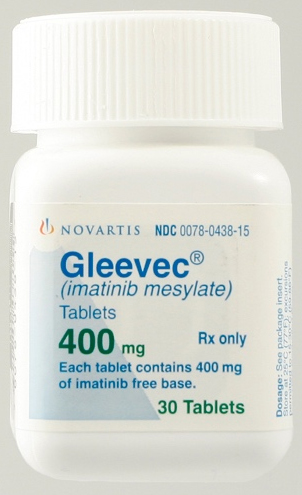
Hình 2: Cấu trúc của Imatinib (Glivec® hay Gleevec®) [16] Hình 3: Thuốc Gleevec® [14]
Các thuốc họ TKIs khác đang có trên thị trường
a. Iressa® (Gefitinib) do hãng AstraZeneca (Thuỵ Điển) sản xuất, sử dụng trong điều trị ung thư phổi không tế bào nhỏ (Non-Small Cell Lung Cancer - NSCLC )
Ung thư phổi là loại ung thư có tỉ lệ mắc bệnh và gây tử vong ở nam giới cao nhất thế giới hiện nay. Trong đó, ung thư phổi không tế bào nhỏ chiếm đến 85% [28]. Phương pháp điều trị ban đầu là dùng thuốc hoá trị thuộc họ platin bằng đường truyền tĩnh mạch [29]. Ở giai đoạn 2, có thể dùng docetaxel cũng bằng đường tĩnh mạch [30,31]. Tuy nhiên các thuốc này không tấn công tế bào ung thư một cách chọn lọc, có nhiều tác dụng phụ và tác động cả đến các tế bào lành [32,33].
Các nghiên cứu lâm sàng đã chứng minh rằng phần lớn ung thư phổi không tế bào nhỏ có liên quan đến sự gia tăng số lượng đột biến của một enzym có tên gọi Epidermal Growth Factor Receptor – Tyrosine Kinase (EGFR-TK) thuộc họ TKs [34]. Khi mang đột biến, enzym này có khả năng tự kích hoạt và từ đó khởi động một chuỗi các phản ứng dây chuyền nhằm kích thích sự tăng trưởng và phân chia của các tế bào ung thư phổi [35]. Do đó, việc ức chế enzym này trở thành một mục tiêu quan trọng trong việc điều trị bệnh ung thư phổi không tế bào nhỏ.
Thuốc Iressa® (Gefitinib, dẫn xuất quinazolin, Hình 4 và 5) được phát triển bởi hãng dược phẩm AstraZeneca đã chứng minh được tác dụng ức chế enzym EGFR-TK. Gefitinib được bắt đầu thử nghiệm lâm sàng pha I vào năm 1997 nhằm đánh giá độ an toàn và dược động học trên nhóm bệnh nhân và người tình nguyện. Thuốc có tác dụng ức chế tốt enzym EGFR ngay cả ở liều thấp hơn nhiều so với liều gây độc. Gefitinib tiếp tục được thử nghiệm lâm sàng pha II với tên gọi IDEAL1 và IDEAL2. Tỉ lệ sống sau 1 năm điều trị của Gefitinib là 29,9% so với 5,5% ở các bệnh nhân dùng thuốc hoá trị liệu. Vì vậy, thuốc được cấp phép sử dụng ở Nhật Bản năm 2002 và ở Hoa Kỳ bởi FDA năm 2003 [36,37]. Thuốc dùng đường uống với liều 250 mg hoặc 500 mg, 1 lần/ngày [38] và có thể dùng riêng lẻ hoặc kết hợp với các loại thuốc khác bằng đường truyền tĩnh mạch [39]. Bên cạnh đó, Gefitinib cũng đang được nghiên cứu lâm sàng cho vài loại ung thư khác như ung thư đầu, cổ, ung thư đại tràng, và ung thư vú [36,39].
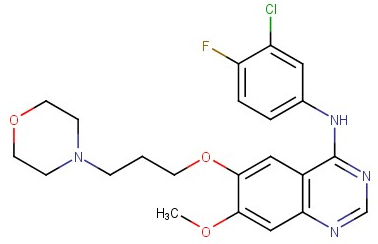

Hình 4: Cấu trúc của Gefitinib [36] Hình 5: Thuốc Iressa® [37]
b. Sutent® (Sunitinib, hãng Pfizer) dùng trong bệnh ung thư biểu mô thận (Renal Carcinoma) và ung thư dạ dày-ruột (Gastrointestinal Stromal Tumors)
Theo thống kê tại Mỹ, trong tổng số ca ung thư thận thì ung thư biểu mô thận chiếm 85% [40]. Điều trị bằng thuốc hoá trị truyền qua đường tĩnh mạch không mang lại hiệu quả cao (chỉ từ 4 – 6%) do sự kháng thuốc mạnh trong các tế bào cầu thận [41]. Một phương pháp điều trị khác mang lại hiệu quả khả quan hơn là dùng các thuốc điều hoà miễn dịch như interferon-alpha, interkeukin-2, hoặc có thể dùng phương pháp cấy tế bào gốc [42,43]. Qua nghiên cứu cho thấy, đặc trưng của bệnh ung thư biểu mô thận là sự gia tăng số lượng vượt trội của một loại protein thuộc họ TKs có tên gọi là Vascular Endothelial Growth Factor Receptor - isoform 2 (VEGFR-2) [44]. Trong các tế bào ung thư, VEGFR-2 kích thích sự tăng trưởng tế bào và tạo mạch máu mới (Angiogenesis) để nuôi sống tế bào ung thư [45]. Do đó, việc ức chế protein này mang lại triển vọng cao trong việc điều trị ung thư biểu mô thận.
Bệnh ung thư dạ dày-ruột hình thành do sự gia tăng số lượng đột biến của mô trung bào trong cấu tạo dạ dày-ruột [46]. Phương pháp điều trị chủ yếu là phẫu thuật cắt bỏ các mô tế bào ung thư. Giống như ung thư biểu mô thận, ung thư dạ dày-ruột cũng có đặc trưng là sự gia tăng số lượng đột biến của một loại protein thuộc họ TKs có tên gọi là c-KIT [47]. Protein đột biến này có khả năng tự kích hoạt nhằm kích thích sự tăng trưởng và phân chia tế bào ung thư một cách nhanh chóng [48]. Do đó, ức chế protein này cũng là một hướng đi khả quan trong điều trị bệnh ung thư dạ dày-ruột.
Thuốc Sutent® (Sunitinib, dẫn xuất chứa vòng indolinamide-pyrrol, Hình 6 và 7) do hãng dược phẩm Pfizer sản xuất được chỉ định dùng trong trường hợp bệnh ung thư biểu mô thận và cả ung thư dạ dày-ruột [49]. Sunitinib có khả năng ức chế protein VEGFR-2 và c-KIT, do đó ức chế sự tăng trưởng, phân chia, và tạo mạch máu mới ở các mô tế bào ung thư [50].
Thử nghiệm lâm sàng trên bệnh nhân ung thư biểu mô thận: Thử nghiệm lâm sàng pha I được tiến hành trên 28 bệnh nhân vào năm 2006. Kết quả đã xác định được liều gây độc ở mức ≥ 75 mg/ngày. Vì thế, Sunitinib được thử nghiệm lâm sàng với liều 50 mg/ngày [51]. Trong thử nghiệm lâm sàng pha II, thời gian điều trị trung bình của 63 bệnh nhân tham gia là 9 tháng. Kết quả là bệnh nhân kéo dài thời gian không tiến triển bệnh (progression-free) được 8,7 tháng, tốt hơn rất nhiều nếu so với 2,4 tháng khi điều trị bằng thuốc điều hoà miễn dịch [52]. Trong thử nghiệm lâm sàng pha III, 750 người bệnh được chọn ngẫu nhiên và chia thành 2 nhóm bằng nhau để so sánh tác dụng của Sunitinib so với thuốc điều hoà miễn dịch (interferon-alpha). Kết quả là bệnh nhân dùng thuốc Sunitinib làm chậm được sự tiến triển bệnh 11 tháng so với 5 tháng khi dùng thuốc interferon-alpha [53].
Thử nghiệm lâm sàng trên bệnh nhân ung thư dạ dày-ruột: Thử nghiệm lâm sàng pha I/II được thực hiện trên 97 bệnh nhân ung thư dạ dày-ruột ở nhiều trung tâm khác nhau. Kết quả là bệnh nhân kéo dài thời gian không tiến triển bệnh khoảng 8 tháng và kéo dài thời gian sống thêm khoảng 20 tháng [54]. Do đó, thử nghiệm lâm sàng pha III được tiến hành trên 312 bệnh nhân, chia làm 2 nhóm: nhóm 1 gồm 207 người sử dụng thuốc Sunitinib và nhóm 2 gồm 105 người không sử dụng thuốc. Kết quả là nhóm sử dụng thuốc kéo dài thời gian không tiến triển bệnh thêm được 6,3 tháng so với 1,5 tháng của nhóm không dùng thuốc [50].
Nhờ vào các kết quả thử lâm sàng mà Sunitinib là thuốc đầu tiên được cấp phép bởi FDA cho điều trị cả 2 loại ung thư nói trên. Thuốc được khuyên dùng với liều 50 mg, 1 lần/ ngày trong vòng 4 tuần, sau đó tạm dừng 2 tuần trước khi bắt đầu đợt điều trị 4 tuần tiếp theo. Các tác dụng phụ được ghi nhận gồm có: viêm da, mệt mỏi, và tăng lipid (mỡ) máu.
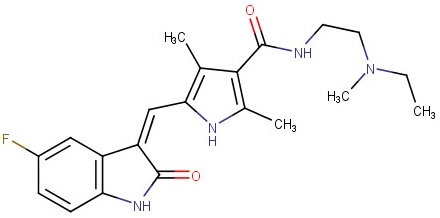
Hình 6: Cấu trúc của Sunitinib [49] Hình 7: Thuốc Sutent® [55]
Các hợp chất khác thuộc họ TKIs đang được thử nghiệm lâm sàng
a. Canertinib
Phần lớn các loại ung thư mô cứng (solid tumor) như ung thư phổi, ung thư thực quản, ung thư vú, ung thư tuyến tiền liệt, và ung thư não có liên quan đến sự gia tăng số lượng bất thường của một họ protein là erbB (còn gọi là họ HER). Đây là một họ protein nằm trong họ TKs, gồm có 4 loại chính là erbB1(HER1), erbB2(HER2/neu), erbB3(HER3), và erbB4(HER4). Các protein này có vai trò quan trọng trong quá trình phát triển và phân chia của các tế bào ung thư [56, 57]. Do đó, việc ức chế hoạt động của họ protein này trở thành mục tiêu của nhiều nghiên cứu. Một trong số đó, hoạt chất CI-1033 (Canertinib, dẫn xuất quinazolin, Hình 8) của Pfizer bước đầu mang lại những hiệu quả tích cực. Canertinib có khả năng ức chế cả 4 protein trong họ erbB đồng thời không có tác động gì lên các protein khác có cấu trúc gần tương tự trong họ TKs [58]. Do đó, tác dụng phụ được ghi nhận chủ yếu là tiêu chảy và phát ban. Canertinib được dùng bằng đường uống, giúp người bệnh không cần điều trị nội trú. Hiện tại, Canertinib được thử nghiệm lâm sàng pha II trên bệnh nhân ung thư phổi không tế bào nhỏ [59] và bệnh nhân ung thư vú di căn [60].
Trong thử nghiệm lâm sàng pha II của canertinib trên 163 bệnh nhân ung thư phổi không tế bào nhỏ đã trải qua hoá trị liệu với thuốc họ platin và không đạt hiệu quả mong muốn, tỉ lệ đáp ứng thuốc của canertinib thấp (khoảng 4%) và tỉ lệ phát triển bệnh sau 26 tuần điều trị cao. Do đó, quá trình nghiên cứu lâm sàng không được tiếp tục [59].
Ngoài ra, Canertinib còn được thử nghiệm lâm sàng pha II trên 198 bệnh nhân ung thu vú di căn với tuổi đời trung bình là 55,5 tuổi. Phần lớn bệnh nhân đều đã trải qua phẫu thuật cắt bỏ mô ung thư, hoá trị liệu, và xạ trị. Các bệnh nhân trong thử nghiệm lâm sàng cũng đã được chẩn đoán có tế bào ung thư di căn sang gan, phổi, và mạch bạch huyết. Kết quả thu được là tỉ lệ sống sau 1 năm điều trị chỉ vào khoảng 4% ở cả ba nhóm. Tỉ lệ này không đủ để tiếp tục nghiên cứu lâm sàng giai đoạn tiếp theo [60].
Tóm lại, các kết quả thu được ban đầu chưa thật sự khả quan và chưa cho phép tiến hành giai đoạn tiếp theo của quá trình nghiên cứu lâm sàng đối với Canertinib. Vì thế, các nhà nghiên cứu cần phải thiết kế lại cấu trúc phân tử của Canertinib cũng như lựa chọn cách đưa hoạt chất này vào cơ thể nhằm cải thiện hiệu quả trong tương lai.
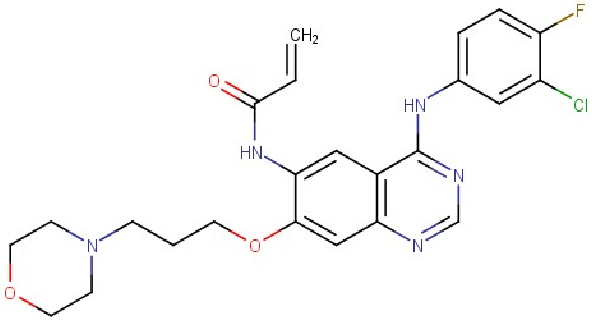
Hình 8: Cấu trúc của Canertinib (CI-1033) [58]
b. BIBW2992
Một trong những vấn đề dẫn đến điều trị bệnh ung thư phổi không tế bào nhỏ không đạt hiệu quả như mong đợi là sự kháng thuốc diễn ra mạnh do đột biến gen trong tế bào ung thư. Sự đột biến này có thể do người bệnh hút thuốc lá, tuy nhiên cũng có thể xảy ra ở người bệnh mắc bệnh ung thư do các tế bào carcinom tuyến (adenocarcinoma), người bệnh gốc châu Á, và nữ giới [61]. Đột biến tác động đến 2 protein quan trọng trong điều trị là EGFR và HER2 (tên gọi khác của protein erbB2) [62]. Từ đó, bệnh nhân trở nên kháng các loại thuốc điều trị hiện có trên thị trường như Erlotinib và Gefitinib [63].
BIBW2992 (dẫn xuất quinazolin, Hình 9) hiện đang được tiến hành thử nghiệm cận lâm sàng (pre-clinical) nhằm thay thế Erlotinib và Gefitinib trong trường hợp bệnh nhân có đột biến ở các gen mã hóa hai protein EGFR và HER2 [64]. BIBW2992 cũng được nghiên cứu trên một số loại ung thư mô cứng khác. Thử nghiệm lâm sàng pha I được tiến hành trên 38 bệnh nhân (19 nam và 19 nữ) với tuổi đời trung bình là 58 tuổi. Kết quả là đã xác định được liều dùng an toàn cao nhất ở mức 70 mg/ngày, theo phác đồ 2 tuần điều trị và 2 tuần tạm nghỉ. Các tác dụng phụ được ghi nhận chủ yếu là tiêu chảy và phát ban [65].
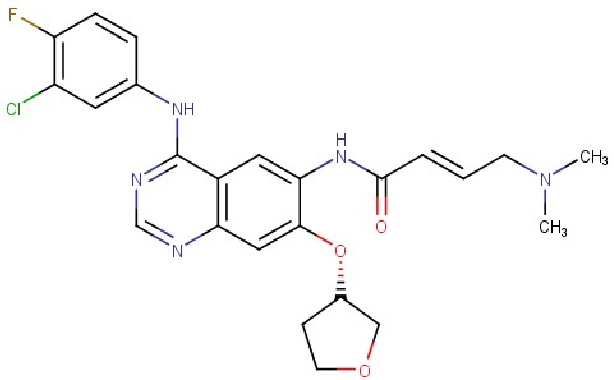
Hình 9: Cấu trúc của BIBW2992 [65]
Tổng kết
Cùng với sự phát triển của kỹ thuật nghiên cứu gen, tế bào học, và liệu pháp miễn dịch, các thuốc kháng ung thư bằng con đường ức chế enzym Tyrosine Kinase nói riêng và các họ enzym khác bằng đường uống mở ra hướng mới trong điều trị ung thư. Dù xuất hiện các cơ chế kháng thuốc nhưng Imatinib vẫn là lựa chọn đầu tiên của họ TKIs trong điều trị ung thư bạch cầu dòng tuỷ mạn tính. Gefitinib được dùng bên cạnh các thuốc hoá trị nhằm tăng hiệu quả điều trị ung thư phổi không tế bào nhỏ. Sunitinib là thuốc đầu tiên được cấp phép cho cả hai loại ung thư: biểu mô thận và dạ dày – ruột. Các hoạt chất đang trong giai đoạn thử nghiệm lâm sàng dù chưa đạt được kết quả như mong muốn nhưng hứa hẹn sẽ được cải thiện để đáp ứng nhu cầu điều trị của bệnh nhân trong tương lai. Hi vọng rằng sự kết hợp giữa các phương pháp trị liệu khác nhau có thể giúp kéo dài hơn nữa thời gian sống, chất lượng sống của bệnh nhân, và tiến tới tiêu diệt triệt để tế bào ung thư.
Về tác giả:
Đinh Châu Phi là sinh viên Cao học, chuyên ngành Dược học tại Khoa Dược, Đại học Angers, Pháp. Phi hiện đang thực tập tại Viện Ung Thư Tây Bắc nước Pháp, tại đây Phi phụ trách kiểm nghiệm thuốc hoá trị và phát triển phương pháp kiểm nghiệm hỗn hợp thuốc giảm đau tiêm vào tuỷ sống.
Võ Đức Duy là nghiên cứu viên sau tiến sĩ (postdoctoral researcher), chuyên ngành Hóa Dược làm việc tại đại học Umeå, Thụy Điển. TS. Duy có nhiều kinh nghiệm trong khám phá và phát triển thuốc như: thiết kế các hợp chất ức chế họ proteins Bcl-2 để khôi phục lại tiến trình tự tử (apoptosis) ở các tế bào ung thư; thiết kế các hợp chất ức chế quá trình tạo ra các miRNA tiền ung thư; thiết kế các hợp chất ảnh hưởng đến tính chất tế bào gốc của tế bào ung thư để khôi phục lại hiệu quả của các loại thuốc hóa trị khi sử dụng kèm ở các tế bào kháng thuốc; và thiết kế các hợp chất kháng sinh thế hệ mới cho vi khuẩn gram âm bằng việc ức chế kim tiêm gây độc tế bào chủ (type III secretion system) nhằm hạn chế sự kháng thuốc trong tương lai. TS. Duy đã có nhiều công trình nghiên cứu trong lĩnh vực hóa dược được đăng tải trên các tạp chí khoa học quốc tế có uy tín.
Biên tập viên: Lê Hoàng
Tài liệu tham khảo
1. Gschwind A, Fischer OM, Ullrich A (2004) The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4(5):361–370.
2. Robinson DR, Wu Y-M, Lin S-F (2000) The protein tyrosine kinase family of the human genome. Oncogene 19(49):5548–5557.
3. Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103(2):211–225.
4. Paul MK, Mukhopadhyay AK (2004) Tyrosine kinase-role and significance in cancer. Int J Med Sci 1(2):101–115.
5. Lemmon MA, Schlessinger J (2010) Cell Signaling by Receptor Tyrosine Kinases. Cell 141(7):1117–1134.
6. Ségaliny AI, Tellez-Gabriel M, Heymann M-F, Heymann D (2015) Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J Bone Oncol 4(1):1–12.
7. Quintanal-Villalonga A, Paz-Ares L, Ferrer I, Molina-Pinelo S (2016) Tyrosine Kinase Receptor Landscape in Lung Cancer: Therapeutical Implications. Dis Markers 2016:1–14.
8. Scheijen B, Griffin JD (2002) Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene 21(21):3314–3333.
9. Faderl S, et al. (1999) The biology of chronic myeloid leukemia. N Engl J Med 341(3):164–172.
10. Shawver LK, Slamon D, Ullrich A (2002) Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell 1(2):117–123.
11. Anafi M, Gazit A, Zehavi A, Ben-Neriah Y, Levitzki A (1993) Tyrphostin-Induced Inhibition of p210bcr-abl Tyrosine Kinase Activity Induces K562 to Differentiate. Blood 82(12):3524–3529.
12. Okabe M, et al. (1992) Effect of herbimycin A, an antagonist of tyrosine kinase, on bcr/abl oncoprotein-associated cell proliferations: abrogative effect on the transformation of murine hematopoietic cells by transfection of a retroviral vector expressing oncoprotein P210bcr/abl and preferential inhibition on Ph1-positive leukemia cell growth. Blood 80(5):1330–1338.
13. Carlo-Stella C, et al. (1996) Selection of myeloid progenitors lacking BCR/ABL mRNA in chronic myelogenous leukemia patients after in vitro treatment with the tyrosine kinase inhibitor genistein. Blood 88(8):3091–3100.
14. All you need to know about Glivec Available at: http://www.biospectrumasia.com/biospectrum/analysis/3205/all-glivec [Accessed September 23, 2016].
15. Druker BJ, et al. (2001) Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 344(14):1038–1042.
16. Deininger M, Buchdunger E, Druker BJ (2005) The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 105(7):2640–2653.
17. Schiffer CA (2007) BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N Engl J Med 357(3):258–265.
18. O’Hare T, et al. (2005) In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res 65(11):4500–4505.
19. Baccarani M, et al. (2009) Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 27(35):6041–6051.
20. Baccarani M (2006) Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 108(6):1809–1820.
21. Radich JP, Gotlib J, Reddy VV ChronicMyeloidLeukemia (Version 1.2016). Natl Compr Cancer Netw. Available at: https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf [Accessed November 14, 2016].
22. Redaelli S, et al. (2009) Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol 27(3):469–471.
23. Hochhaus A, et al. (2008) Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia 22(6):1200–1206.
24. Shah NP, et al. (2010) Potent, transient inhibition of BCR-ABL with dasatinib 100 mg daily achieves rapid and durable cytogenetic responses and high transformation-free survival rates in chronic phase chronic myeloid leukemia patients with resistance, suboptimal response or intolerance to imatinib. Haematologica 95(2):232–240.
25. Kantarjian H, et al. (2010) Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 362(24):2260–2270.
26. Kantarjian HM, et al. (2007) Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome–positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 110(10):3540–3546.
27. Kantarjian H, et al. (2006) Nilotinib in imatinib-resistant CML and Philadelphia chromosome–positive ALL. N Engl J Med 354(24):2542–2551.
28. Ettinger DS, et al. (2013) Non–small cell lung cancer, version 2.2013. J Natl Compr Canc Netw 11(6):645–653.
29. Group IALCTC, others (2004) Cisplatin-based adjuvant chemotherapy in patients with completely resected non–small-cell lung cancer. N Engl J Med 2004(350):351–360.
30. Alberti W, et al. (1995) Chemotherapy in Non-Small-Cell Lung-Cancer - a Metaanalysis Using Updated Data on Individual Patients from 52 Randomized Clinical-Trials. Br Med J 311(7010):899–909.
31. Fossella FV, et al. (1995) Phase II study of docetaxel for advanced or metastatic platinum-refractory non-small-cell lung cancer. J Clin Oncol 13(3):645–651.
32. Shepherd FA, et al. (2000) Prospective randomized trial of docetaxel versus best supportive care in patients with non–small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol 18(10):2095–2103.
33. Schiller JH, et al. (2002) Comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N Engl J Med 346(2):92–98.
34. Mendelsohn J (2000) Blockade of receptors for growth factors: an anticancer therapy—the fourth annual Joseph H. Burchenal American Association for Cancer Research Clinical Research Award Lecture. Clin Cancer Res 6(3):747–753.
35. Pao W, Chmielecki J (2010) Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 10(11):760–774.
36. Muhsin M, Graham J, Kirkpatrick P (2003) Gefitinib. Nat Rev Drug Discov 2(7):515–516.
37. Iressa Back in the US for Lung Cancer, Now as First-Line Medscape. Available at: http://www.medscape.com/viewarticle/847853 [Accessed November 17, 2016].
38. Nurwidya F, Takahashi F, Takahashi K (2016) Gefitinib in the treatment of nonsmall cell lung cancer with activating epidermal growth factor receptor mutation. J Nat Sci Biol Med 7(2):119.
39. Herbst RS, Fukuoka M, Baselga J (2004) Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer 4(12):979–987.
40. Cohen HT, McGovern FJ (2005) Renal-cell carcinoma. N Engl J Med 353(23):2477–2490.
41. Yagoda A, Abi-Rached B, Petrylak D (1995) Chemotherapy for advanced renal-cell carcinoma: 1983-1993. Semin Oncol 22(1):42–60.
42. Yang JC (2003) Randomized Study of High-Dose and Low-Dose Interleukin-2 in Patients With Metastatic Renal Cancer. J Clin Oncol 21(16):3127–3132.
43. Fisher RI, Rosenberg SA, Fyfe G (2000) Long-term survival update for high-dose recombinant interleukin-2 in patients with renal cell carcinoma. Cancer J Sci Am 6 Suppl 1:S55-57.
44. Rini BI (2005) VEGF-targeted therapy in metastatic renal cell carcinoma. The Oncologist 10(3):191–197.
45. Ferrara N, Davis-Smyth T (1997) The biology of vascular endothelial growth factor. Endocr Rev 18(1):4–25.
46. Joensuu H (2006) Gastrointestinal stromal tumor (GIST). Ann Oncol 17(suppl 10):x280–x286.
47. Rammohan A (2012) A gist of gastrointestinal stromal tumors: A review. World J Gastrointest Oncol 5(6):102.
48. Rubin BP, et al. (2001) KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 61(22):8118–8121.
49. DrugBank ed. (2016) Sunitinib. DrugBank. Available at: http://www.drugbank.ca/drugs/DB01268 [Accessed November 14, 2016].
50. Le Tourneau C, Raymond E, Faivre S, others (2007) Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther Clin Risk Manag 3(2):341.
51. Faivre S (2006) Safety, Pharmacokinetic, and Antitumor Activity of SU11248, a Novel Oral Multitarget Tyrosine Kinase Inhibitor, in Patients With Cancer. J Clin Oncol 24(1):25–35.
52. Motzer RJ, et al. (2006) Sunitinib in patients with metastatic renal cell carcinoma. Jama 295(21):2516–2524.
53. Motzer RJ, et al. (2006) Phase III randomized trial of sunitinib malate (SU11248) versus interferon-alfa (IFN-α) as first-line systemic therapy for patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol 24(90180):LBA3-LBA3.
54. Reichardt P, et al. (2016) Correlation of KIT and PDGFRA mutational status with clinical benefit in patients with gastrointestinal stromal tumor treated with sunitinib in a worldwide treatment-use trial. BMC Cancer 16(1). doi:10.1186/s12885-016-2051-5.
55. Mail BFH for the D (2010) Mother denied cancer drugs that were promised by Labour says, “I just want to see my sons grow up.” Mail Online. Available at: http://www.dailymail.co.uk/health/article-1264932/Mother-denied-cancer-drugs-promised-Labour-says-I-just-want-sons-grow-up.html [Accessed September 23, 2016].
56. Graus-Porta D, Beerli RR, Daly JM, Hynes NE (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16(7):1647–1655.
57. Olayioye MA (2001) Intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res 3(6):1.
58. Slichenmyer WJ, Elliott WL, Fry DW (2001) CI-1033, a pan-erbB tyrosine kinase inhibitor. Semin Oncol 28(5N):80–85.
59. Janne PA, et al. (2007) Multicenter, Randomized, Phase II Trial of CI-1033, an Irreversible Pan-ERBB Inhibitor, for Previously Treated Advanced Non Small-Cell Lung Cancer. J Clin Oncol 25(25):3936–3944.
60. Rixe O, et al. (2009) A randomized, phase II, dose-finding study of the pan-ErbB receptor tyrosine-kinase inhibitor CI-1033 in patients with pretreated metastatic breast cancer. Cancer Chemother Pharmacol 64(6):1139–1148.
61. Janne PA (2005) Epidermal Growth Factor Receptor Mutations in Non-Small-Cell Lung Cancer: Implications for Treatment and Tumor Biology. J Clin Oncol 23(14):3227–3234.
62. Janne PA (2005) Epidermal Growth Factor Receptor Mutations in Non-Small-Cell Lung Cancer: Implications for Treatment and Tumor Biology. J Clin Oncol 23(14):3227–3234.
63. Riely GJ (2006) Clinical Course of Patients with Non-Small Cell Lung Cancer and Epidermal Growth Factor Receptor Exon 19 and Exon 21 Mutations Treated with Gefitinib or Erlotinib. Clin Cancer Res 12(3):839–844.
64. Li D, et al. (2008) BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27(34):4702–4711.
65. Eskens FALM, et al. (2008) A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer 98(1):80–85.